食品微生物の遺伝子検査の基礎を理解するためには、DNAシーケンサーの原理の理解が不可欠である。本記事では、DNAの配列決定法について、サンガー法(電気泳動、キャピラリーシーケンサー)、次世代シーケンサー(イルミナシーケンサー、Ion Torrent シーケンサー)、第3世代シーケンサー(ナノポアシーケンサー)の各DNAシーケンサーの原理をわかりやすく説明する。原理や仕組みについては、食品微生物学の初心者がイメージとした理解できることを目的として、大まかな原理について、一部、動画もまじえてわかりやすく説明する。また、各シーケンサーの食品微生物学分野の応用例にも触れる。
サンガー法
DNA の配列決定は、英国のフレデリック・サンガー博士により 考案された。 その名前にちなんでサンガー法と呼ばれている。サンガーが博士は、この功績により1980年にノーベル賞を受賞している。ちなみに、サンガー博士は、 DNA の配列決定法を開発する前には、タンパク質の配列決定法を開発している。この功績により1958年にノーベル賞を受賞している。すなわち、生涯にわたって、別々の業績で二度ノーベル賞を受賞しているということになる。
電気泳動法
以下にサンガー法の原理を説明する。
DNA の配列を決定するためにも PCR 反応を使う。もちろん、サンガーが行ったオリジナルの方法ではPCR法がない時代だったので、PCR法ではない。しかし、ここでは、現在でも広く使われているサンガー法の原理に基づくキャピラリースーケンサーの原理を理解するために、あえて、PCR法という言葉を使ったほうが分かりやすいので、PCR法で説明する。
※PCR の仕組みの理解については、下記の記事をご覧ください
PCR法とリアルタイムPCRをわかりやすく説明します
ただし PCR 反応と異なる点は、プライマーを一本しか使わないという点だ。プライマー一本だけからの伸長反応が行われる。つまり、一方通行である。 ただし、これだけでは 、単に DNA が伸長されるだけでその他は何も起きない。

問題はここからである。
このような DNA の伸長反応を行う際に、 DNA 伸長の材料となるヌクレオチドが必要となる。 ヌクレオチドとは、A,T,C,Gの4つの塩基 と糖およびリン酸が結合したものである。要するに DNA を合成する4文字の一つ一つの材料ブロックと考えれば良い。
さてここからがサンガー博士の天才的な発想である。
DNA を伸長する際に各種(A,T,G,C)のヌクレオチドの一部に欠陥ヌクレオチドを混ぜ込んでおく。欠陥ヌクレオチドとは、下の図のように、 3’部分のOH基がHに置換されたものだ。 DNA ポリメラーゼは酵素の基質特異性があるので、3’末端がHになってしまっていてはその構造を認識できない。つまりこのような欠陥ヌクレオチドが入っている場合には DNA ポリメラーゼはその先は伸長できないことになる。
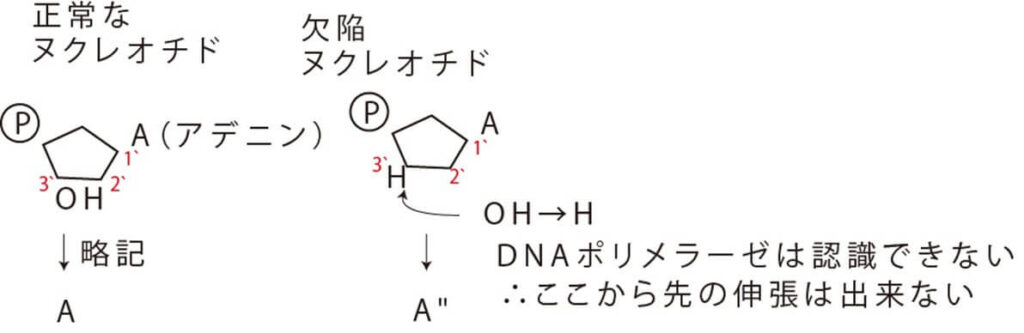
ここで、欠陥ヌクレオチドだけを反応液に入れておけば、 DNA伸長は一塩基伸長したところで、ストップする。それ以上は何も起きない。
ポイントは、大多数は正常なヌクレオチドを入れておき、わずかに欠陥ヌクレオチド混ぜ込んでおくということだ。
このようなことをすると何が起きるだろうか?下の図を見て頂きたい。
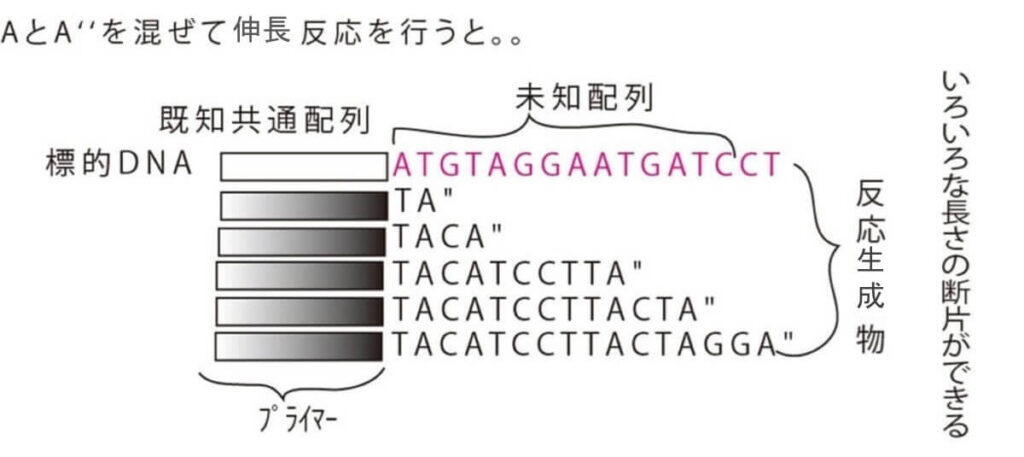
大多数は正常なヌクレオチドなので、多くの場合、順調に伸長が行われる。しかし、ところどころに欠陥ヌクレオチドが入っているために、伸長が停止する断片も生じる。
今、4文字中、Aのみについて、欠陥ヌクレオチドをいれた反応系を考えてみよう。下の図で、 Aの欠陥ヌクレオチドの濃度がわずかだった場合、一番下の長い断片ができる確率が高くなる。一方、Aの欠陥ヌクレオチドの濃度が著しく高い場合、どうなるだろうか? 一番上の一番短い断片が出来る確率が高くなる。
このように Aの欠陥ヌクレオチドの濃度 は低すぎても高すぎてもダメだ。適度に絶妙な配合で欠陥ヌクレオチド混ぜ込んでおくと、下の図のように、色々な長さの断片の伸長産物ができることになる。
これがサンガー法のポイントである。
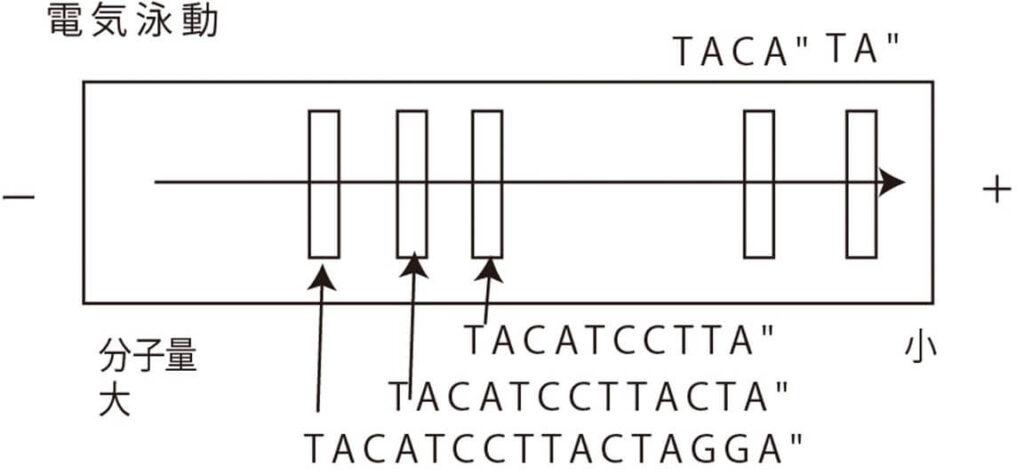
このようにして作られた様々なサイズの DNA 断片は電気泳動で流し、ある一定時間で電気泳動を止めれば、各バンドは異なる移動度で展開される。分子量の小さいTA”は先頭のほうに進み、 分子量の大きいTACATCCTTACTAGGA”は開始点の近くに止まっている。
以上は、4つの塩基の中でAについてのみ、欠陥ヌクレオチドを入れておいた場合のパターンだ。同じように残りの3つの塩基G,C,Tに ついてもそれぞれ欠陥ヌクレオチドを入れておいた反応を別々の反応液で行う。
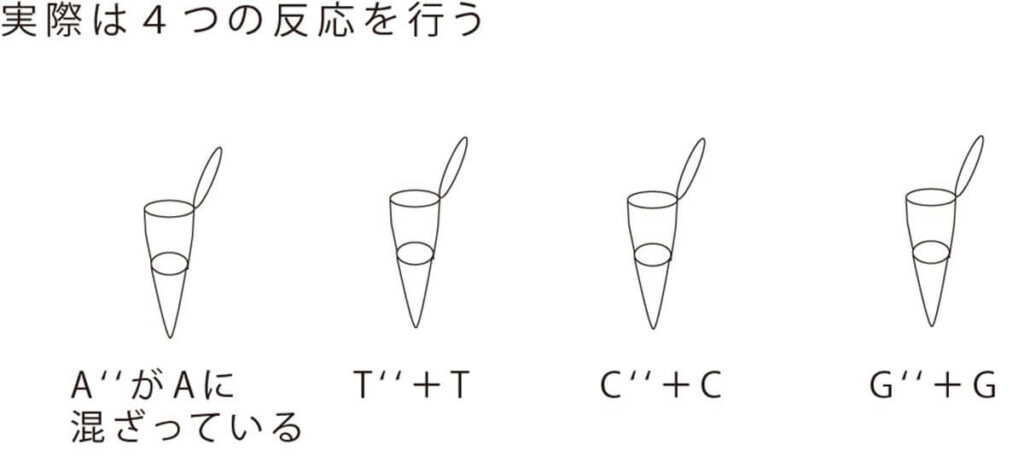
そして4つの反応液を電気泳動の別のレーンにそれぞれ展開させると、下の図のようになる。
※DNAの電気泳動の原理の基礎を確認したい方は、下記記事の電気泳動の部分を確認してください
PCR法とリアルタイムPCRをわかりやすく説明します
電気泳動の4つのレーンにそれぞれ異なるサイズのバンドが展開される。プラス電極側に最も早く進んでいるのが最も小さい断片だ。ここでポイントはATGCの四つのレーンで同じ移動度のバンドは決して生じないということだ。
そして小さい断片順にどのレーンにそれらの断片が配置されているかを読んでいく。
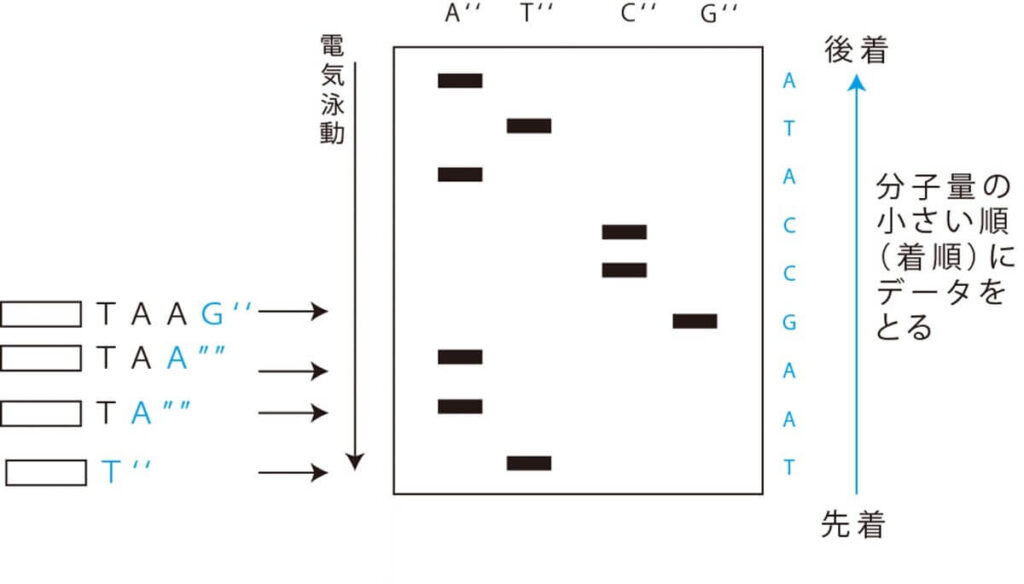
結果として、元々のオリジナル DNA の相補的な DNA 配列がわかる。そして元々のオリジナル DNA の配列は、 その総合的な DNA 配列を裏返せば良いということになる。
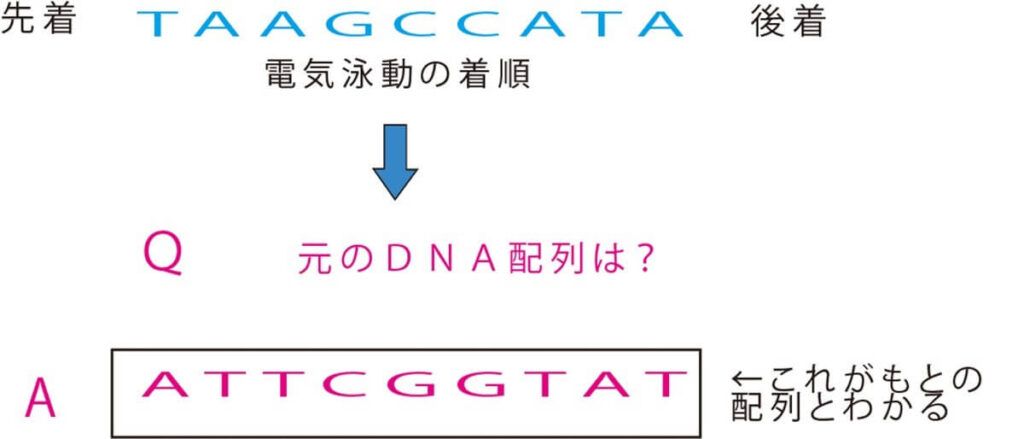
以上が古典的なサンガーの遺伝子決定法の原理だ。 このような原理の理解が現在のすべてのDNAシーケンサーの仕組みの基本となっている。
食品微生物検査と平板ゲル電気泳動型シーケンサー
ところで、上に述べた方法では電気泳動を止めて、そこに存在している DNA 断片を順番に確認していくという作業であった。 1990年代に 初頭にATCGの4つのヌクレオチドにあらかじめ蛍光色素をつけておく方法が開発された。4つのヌクレオチドに別々の蛍光色素をつけておけば、ある一定時間で電気泳動を止めなくてもよい。連続的に電気泳動を流し、順次、終着点に到着する蛍光色素を機械で読み取るだけで済む。1990年代に登場したのが、平板のゲルを用いて電気泳動型蛍光色素DNA シークエンサーである。このタイプのシーケンサーは1990年代には広く普及した。しかし、1990年代後半にキャピラリー型シーケンサーが登場し、その役割を終えた。
このように平板ゲル電気泳動型シーケンサーは、実用的に食品微生物の検査分野で用いられた期間はわずかであった。しかし、このタイプのシーケンサーはサンガー法の原理を学ぶ教育機器としては優れている。実際、筆者は、キャピラリーシーケンサーが登場したあと10年間は、大学の3年生むけの食品微生物学実験において、 平板ゲル電気泳動型蛍光シーケンサー を使っていた。視覚的にサンガー法の原理が理解しやすいので、教育機器としては優れていた。しかし、2012年以降、大学の教育用にもキャピラリーシークエンサーへ完全移行し、筆者の研究室では 平板ゲル電気泳動型シーケンサー の教育機器としての役割も終了した。
キャピラリーシーケンサー法
続けて1990年半ばにはキャピラリー型シークエンサーが登場した。ABI社から販売されたが、キャピラリーシークエンシングの基幹技術は日立製作所の開発によるものである。キャピラリーシークエンサーでは ATCG の4つのひとつの一本のキャピラリーの中にまとめて展開する。4つの塩基には別々の蛍光色素が付いているのでキャピラリーの出口から出てくる様々の大きさな DNA 断片はそれぞれ別の色の蛍光を発していることになる。このシグナルを機械で読み取ることによって DNA の配列がわかる。
キャピラリーシークエンサーのおおまかな原理については下の下のビデオををご覧ください。
食品微生物検査とキャピラリーシーケンサー
キャピラリーシークエンサーは、 1990年後半から現在に至るまで食品微生物の遺伝子による検査における主力として活躍している。現在、後述する次世代シークエンサーは食品微生物分野において特に全ゲノム解析による分子疫学解析や16SrDNAによるメタゲノム解析( 16SrRNAアンプリコンシーケンス )などの活用で急速に普及中ではある。しかし、現時点では、食品微生物分野においては、特定の遺伝子の配列を読むだけだ目的を達する場合も多い。
特に食品から分離した特定の微生物株の16SrDNAによる同定においては、 キャピラリーシークエンサーを用いるのがもっとも便利だ。また、日本では、腸管出血性の分子疫学的解析において、2018年からEHECの血清型O157、O26およびO111による食中毒の感染源特定の分子疫学的解析法として用いられているMLVAもキャピラリーシークエンサーで実施されている。
※分子疫学解析としてのMLVA法の基礎を確認したい方は下記記事のMLVA項目をご覧ください
病原菌や食中菌の感染ルート把握のための分子疫学解析手法(菌株の識別法)のすべてをわかりやすく解説します
次世代シークエンサー
上述したキャピラリーシーケンサーは1990年代後半に出現後、現在にいたるまで、DNAシーケンサーのゴールドスタンダードとして活躍しつづけている。
一方、2007年頃から、これまでのサンガー法とは、根本的に異なる原理にもとづく新しいシーケンス技術が実用的に普及しはじめた。次世代シークケンシング(Next Generation Sequencing、NGS)の登場によりサンガー法に比べて桁違のスピードと低コストでDNA配列を読むことができるようになった。
NGSでは、あらかじめ微生物の全ゲノムを300塩基程度の短いDNA断片に切断する。そして切断された大量のDNA断片の塩基配列を同時にミクロ基盤上で決定する。
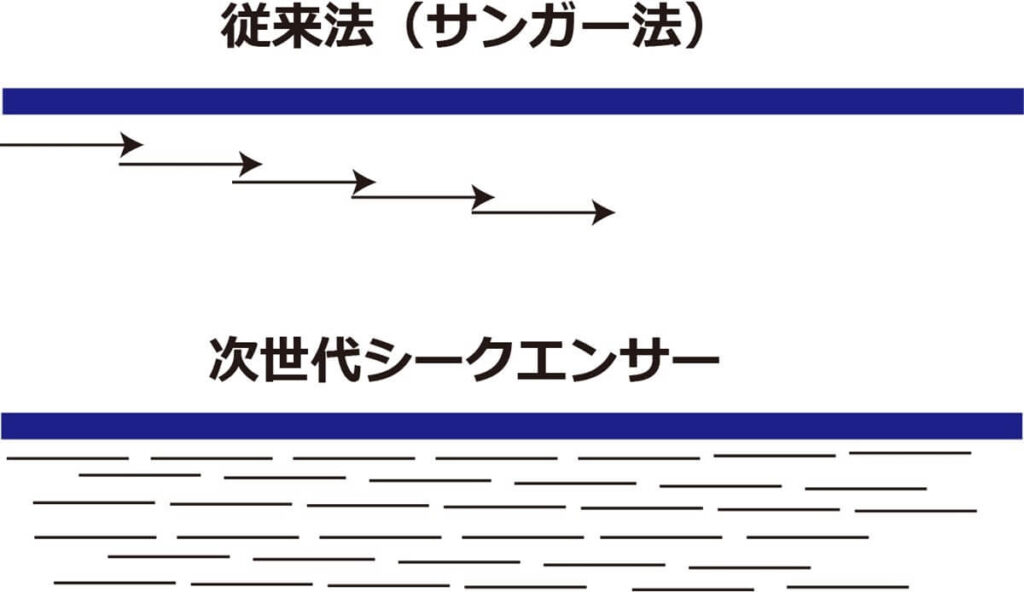
たとえで言うと、これまで数万ページの本を1人で3年間かけて読んでいたのがサンガー法であるならば、NGSはこれらのページを1ページずつバラバラにして数万人がたった1日で読むようなものだ。サンガー法に比べて、数千倍から数万倍の速度で遺伝子配列を決定できるようになった。
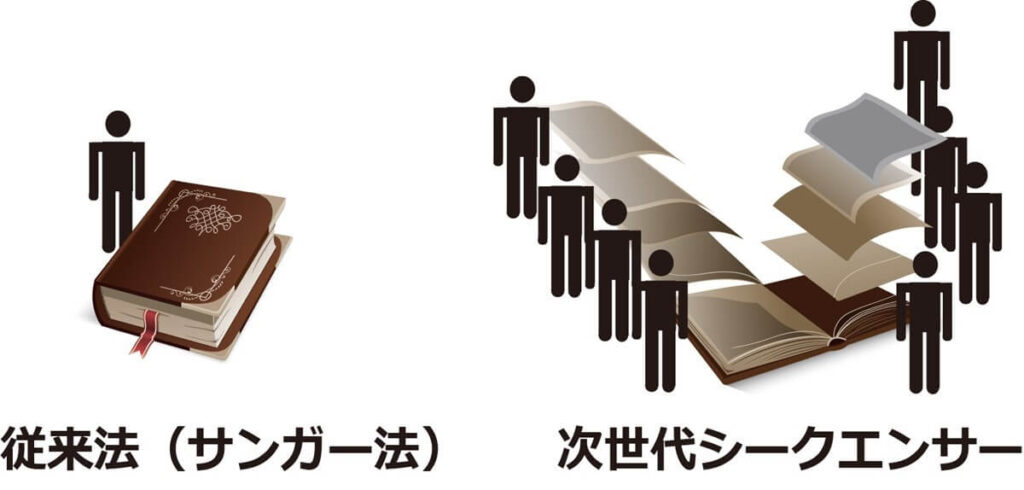
以下に、次世代シークエンサーの代表的な2つのシーケエンサーの原理ついて解説する。ただし、本記事は、あくまでも入門者向けに、大まかな特徴を示すための説明である。したがって、細部は割愛して説明している部分もある。各シークエンサーのさらに詳細な原理を知りたい方は、下記のサイトを参照を薦める。
イルミナ(Illumina) シーケンサ ー
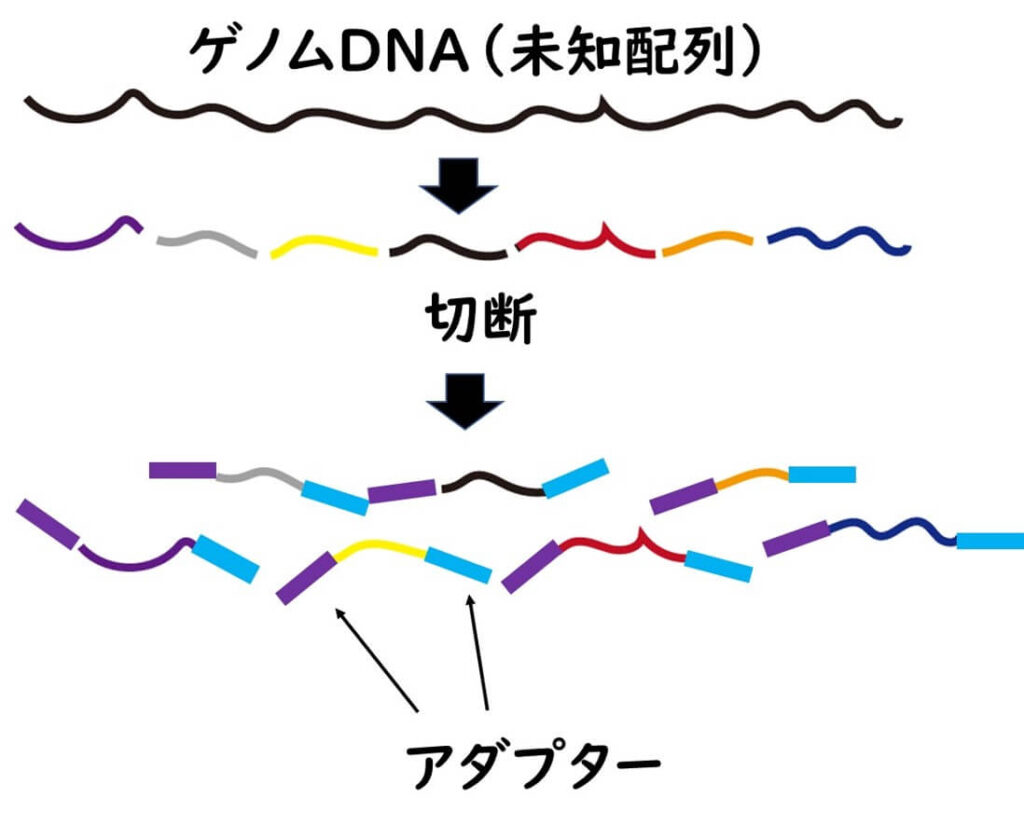
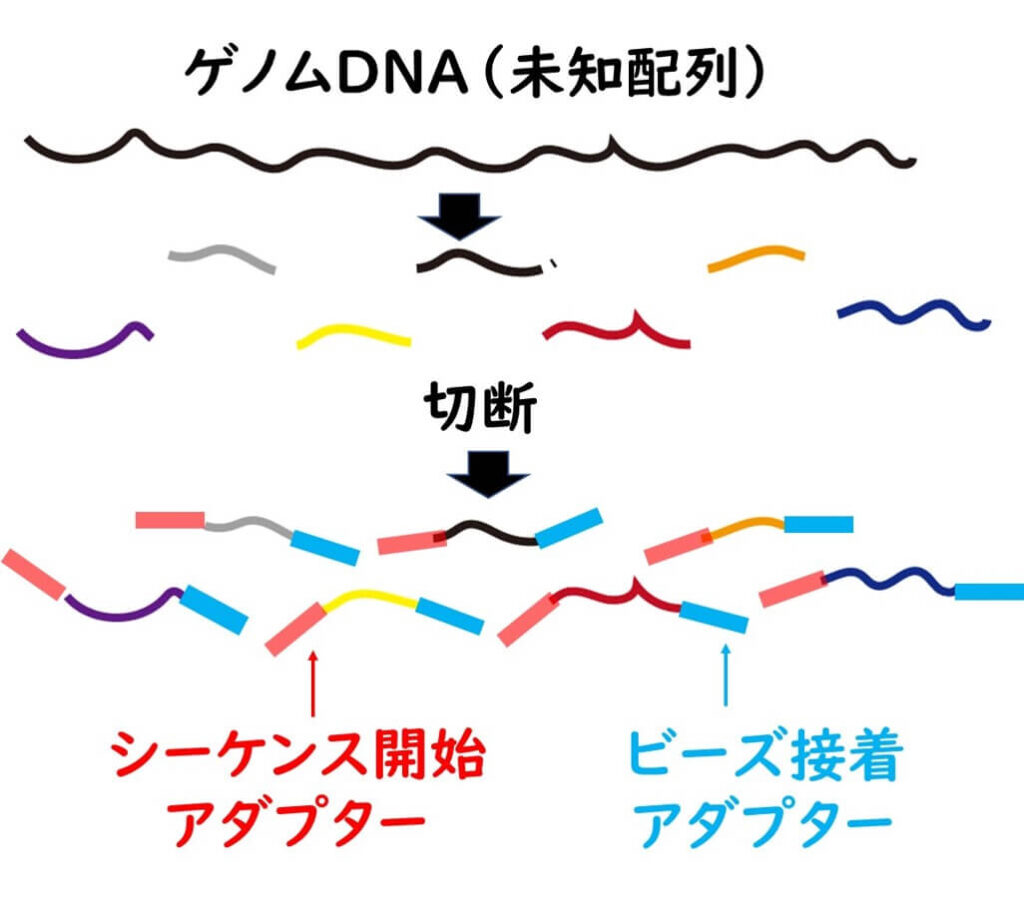
1)まずは対象とする微生物の全ゲノムを制限酵素によって短い断片(200bp程度)にする。
2)バラバラに切断された短いDNA断片の両方の末端にadapter配列を結合させる。
以上の操作は、基本的には、ion Torrentも同じである。
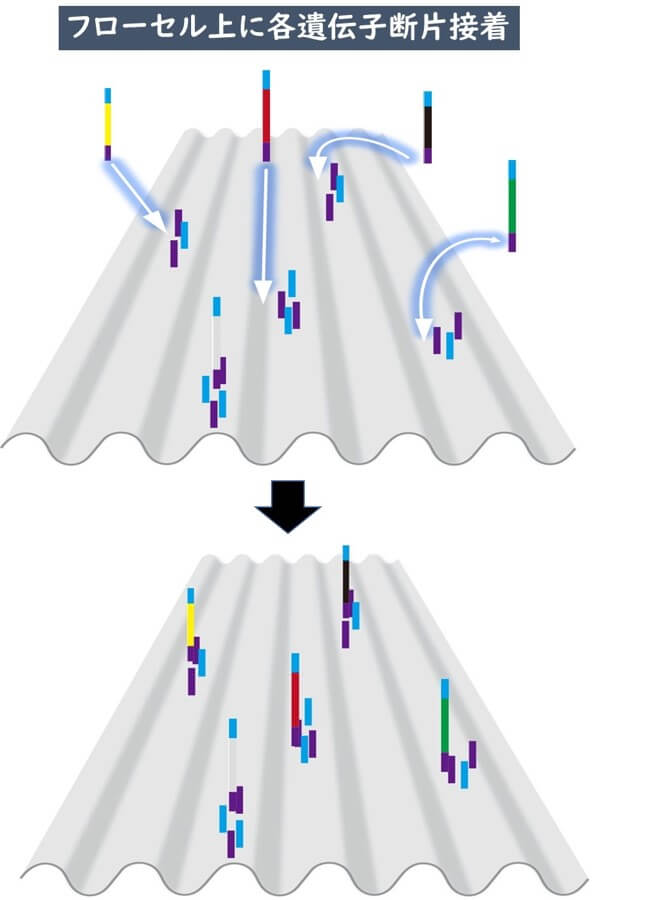
illuminaではフローセル上で標的DNAの各種断片を増幅させる。
1)フローセルの上にあらかじめアダプターの遺伝子配列と相補的な遺伝子配列を持った断片を埋め込んでおく。
2)このフローセルにアダプターと結合済みの標的DNA断片を流す。
3)標的DNAはフローセル上の各所に結合する。
フローセルのアダプターと結合した標的DNA断片を鋳型として相補的な遺伝子断片が合成される。
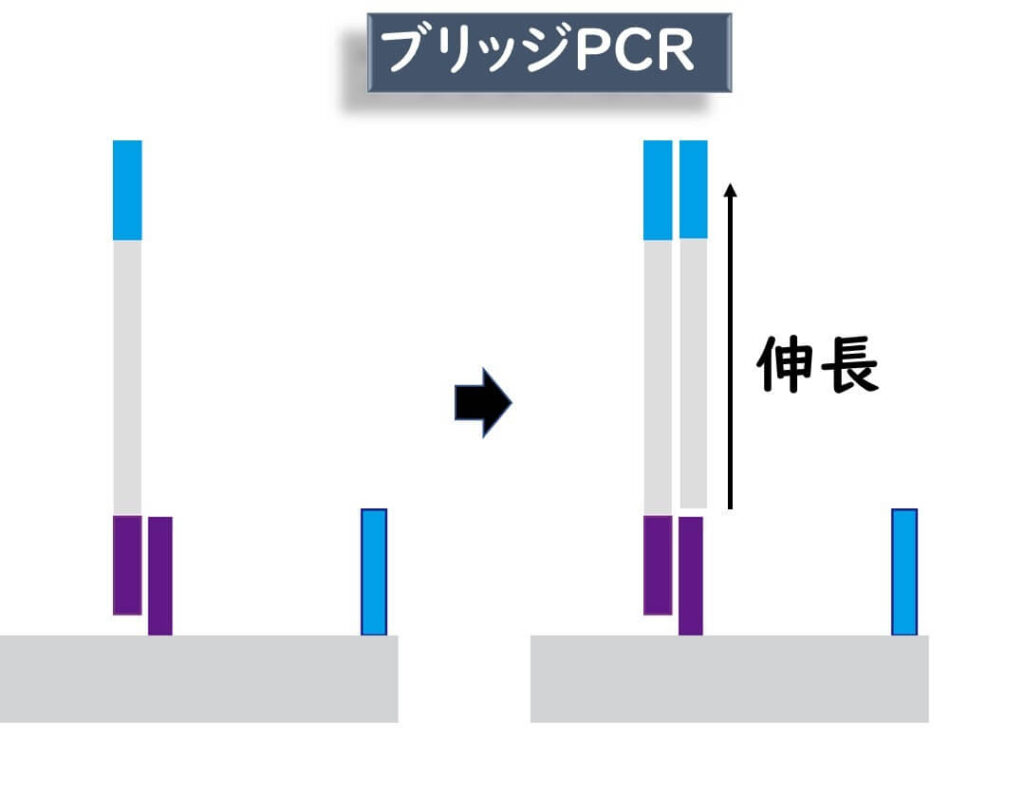
合成された遺伝子断片の末端は近隣に存在している別のadapter配列にに結合する(ブリッジを形成する)。
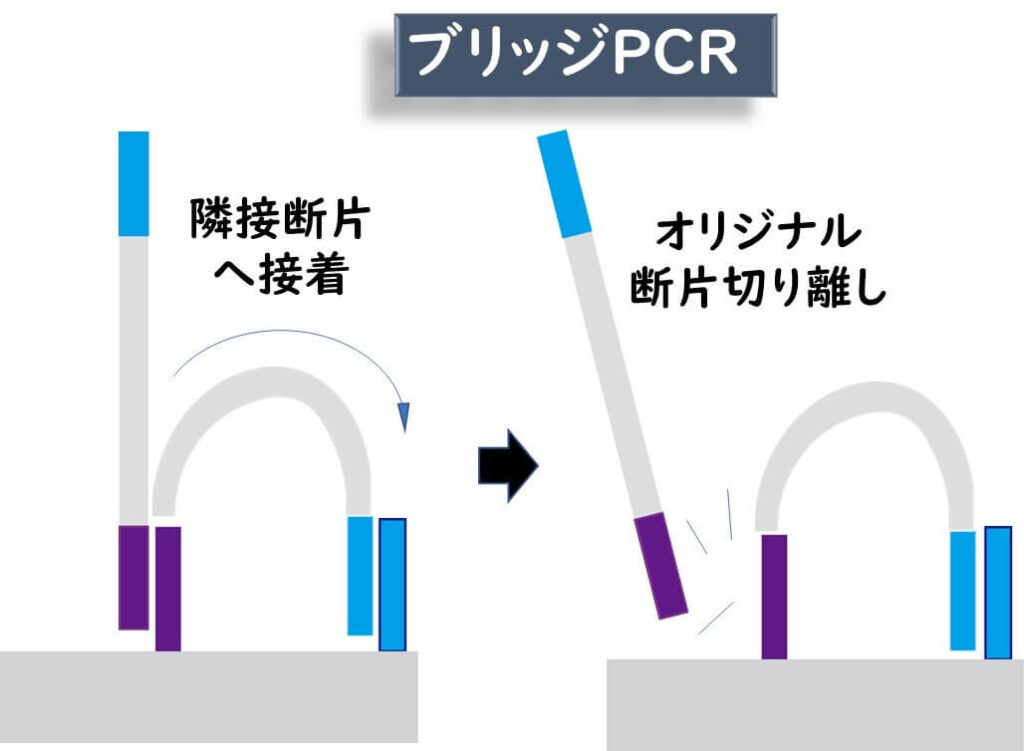
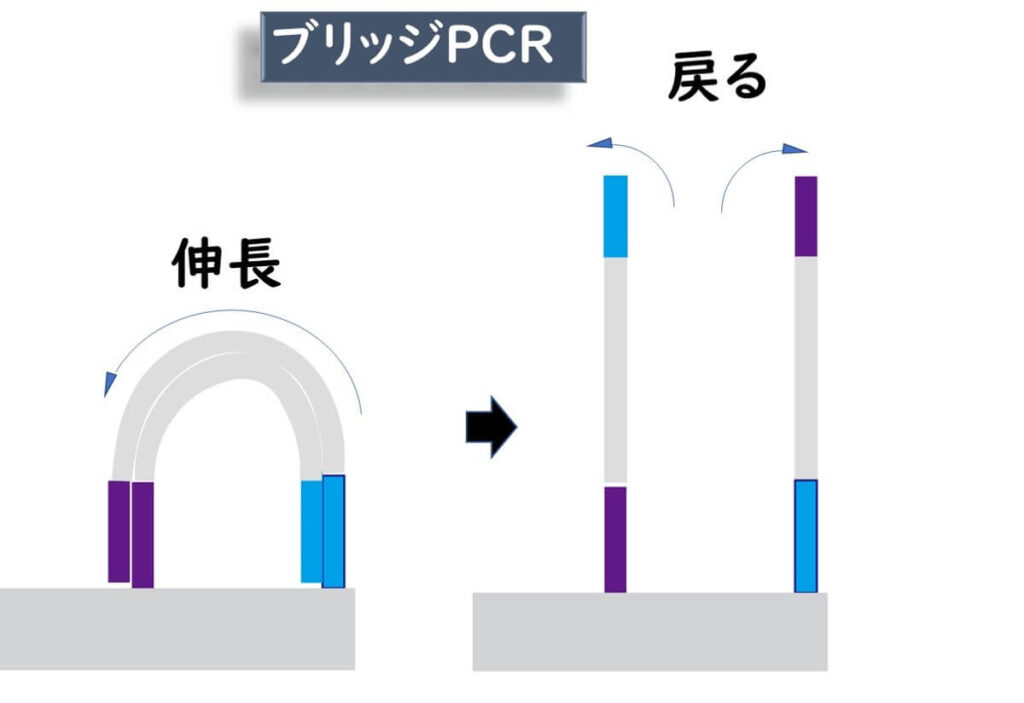
このブリッジを鋳型として、隣のアダプタ配列からさらに伸張反応が起きる。
このような反応が繰り返されていく。
illuminaにおけるPCR増幅をブリッジPCRと呼ぶ。
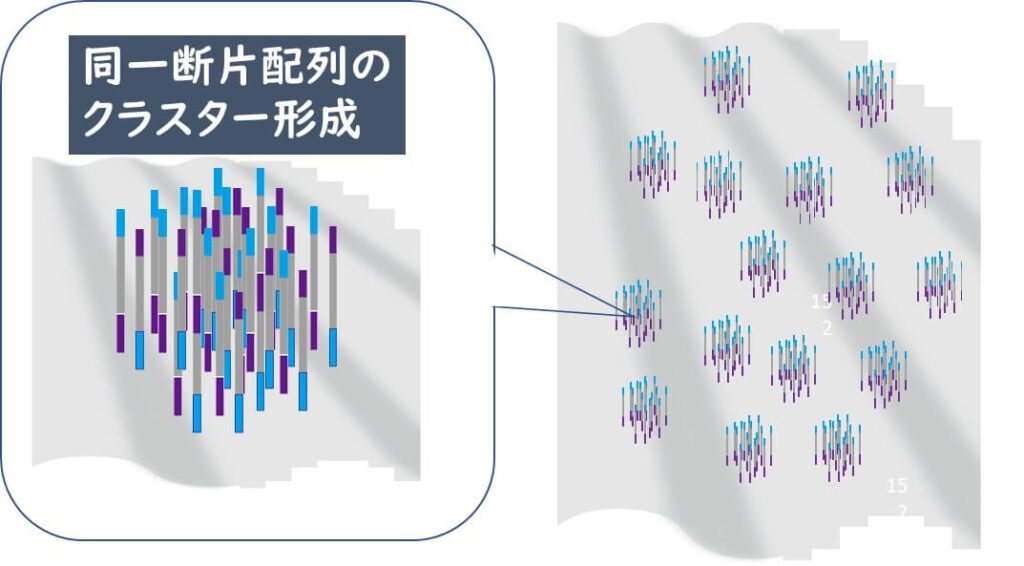
フローセルにはあらかじめ無数のアダプタ配列がクラスターを形成するように配置されてある。それぞれのアダプタ配列の結合した目的の遺伝子断片が大量に束になって増幅されることになる。
もちろん2種類以上のの遺伝子断片が混ざって増幅されてしまう場合もある。しかし、極力そのようにならないように試料の濃度を調節してフローセルに流す。複数の遺伝子断片が混ざったクラスターはその後の遺伝子配列解析段階において混乱した情報を発信しノイズとして排除される。
illuminaによる塩基配列決定の原理は、1塩基毎に異なる蛍光色素を標識しておくことである。この点においては従来のキャピラリー式DNAシークエンサーと似ている。
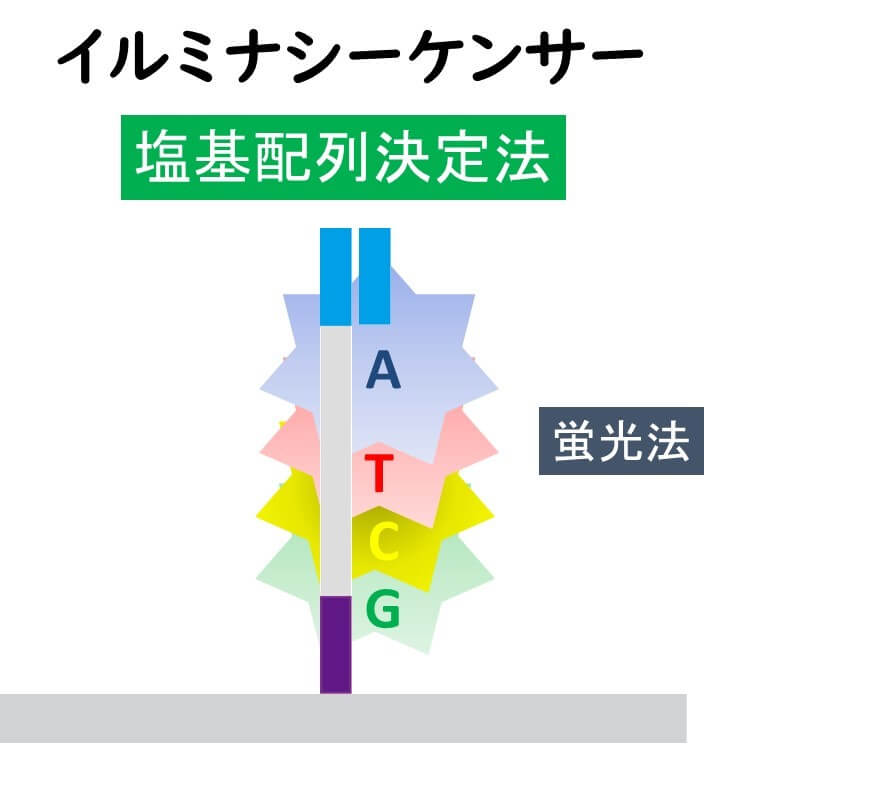
ただし1塩基が伸張される毎に蛍光を測定し、その後その蛍光と塩基伸長のブロックを取りはずす反応行う。そして再度次の伸長を行う。このように丁寧に伸長していくので、塩基の読み間違いが少ない点が特徴。すなわちSNP解析を行うにはとても向いている。ただし、このような反応毎回行うので、試薬の機能の減衰があり、長い配列を読むのが苦手である。
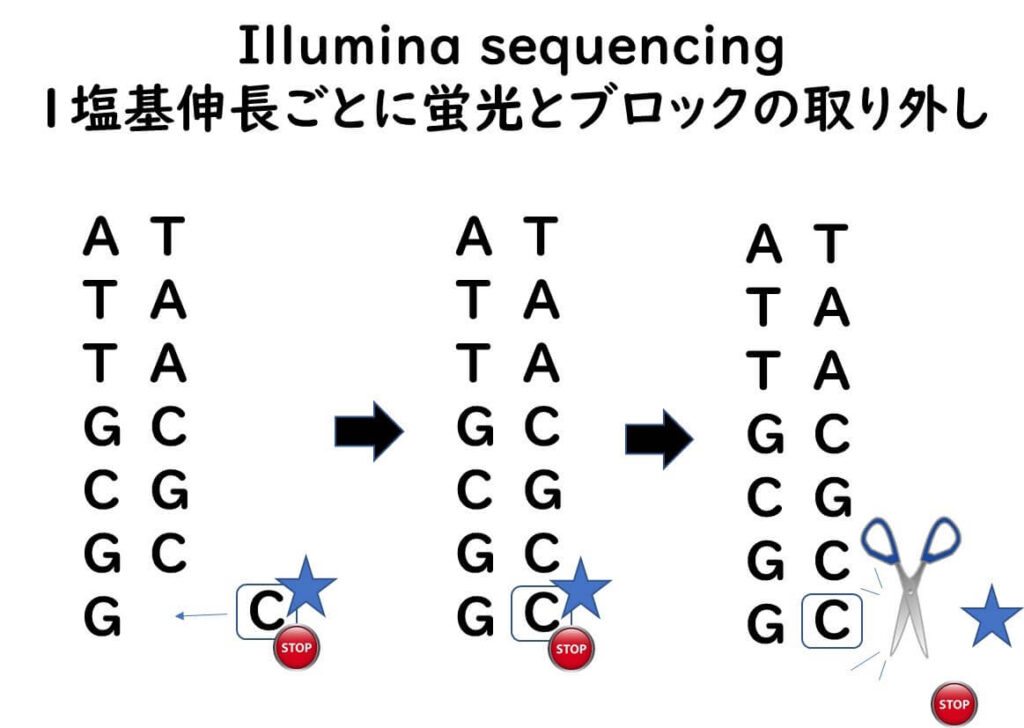
上で述たようなミクロクラスターはフローセル上に何十万というレベルで存在する。
一つ一つのクラスターの遺伝子配列を同時に蛍光標識で読むことが可能である。これにより一挙に大量の遺伝子配列の解析が可能となる。

このようにして塩基配列が決定された無数のショートシークエンスをコンピューター内でつなぎ合わせる作業を行う。

以上のプロセスを下記動画にしてみました。
ion torrent シークエンシング
Ion Torrentにおいてもまず標的DNAを制限酵素により切断し両方の末端にアダプター配列を接続するところまでは、基本的には、illuminaと同様である。
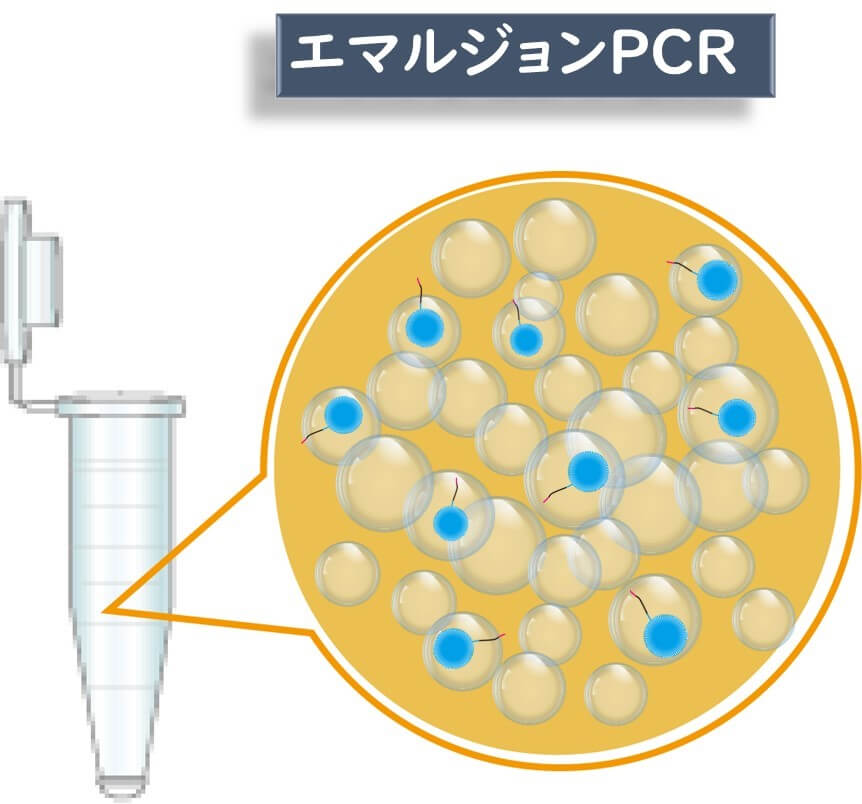
Ion TorrentにおいてはこれらのDNA断片を、マイクロビーズに結合させる。

Ion Torrentにおける目的遺伝子断片の増幅は、エマルジョンPCRによって行う。エマルジョンPCRとは、PCR反応液を油と混合して無数の水滴をエマルジョンの中に作らせるPCRのことである。一つ一つの水滴の中でPCR反応が行われる。これにより小さなマイクロチップの中でも無数のPCR反応を同時に行なえる。
一つ一つ水滴の中に、配列を決定する目的断片1分子のみが入る確率が高くなるように、あらかじめ目的DNA断片の分子量とマイクロビーズのモル比を調整しておく。
ビーズ上にあらかじめ設置してあるアダプタ配列と相補的に接着する。
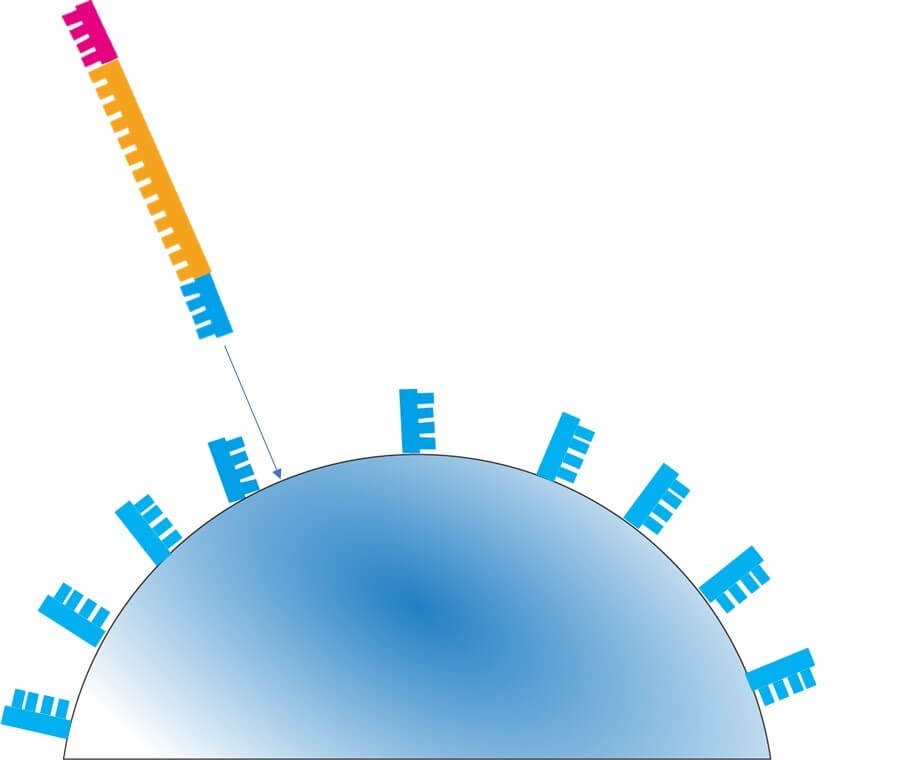
片方のプライマーがビーズ上に接着している点を除けば通常のPCRと同じ反応が行われ、目的断片が増幅されていく。
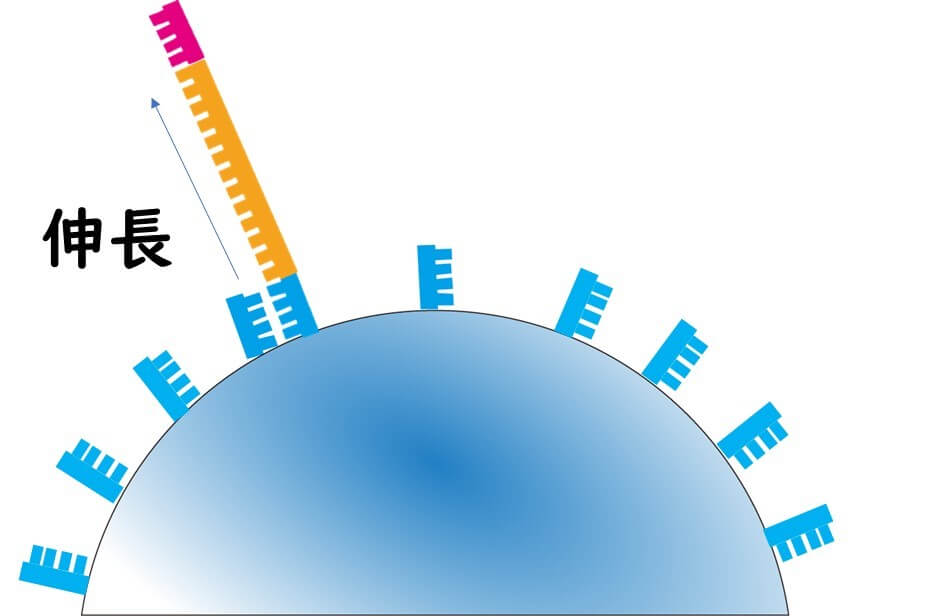
伸長の鋳型に用いた断片は、熱変性により解離する。
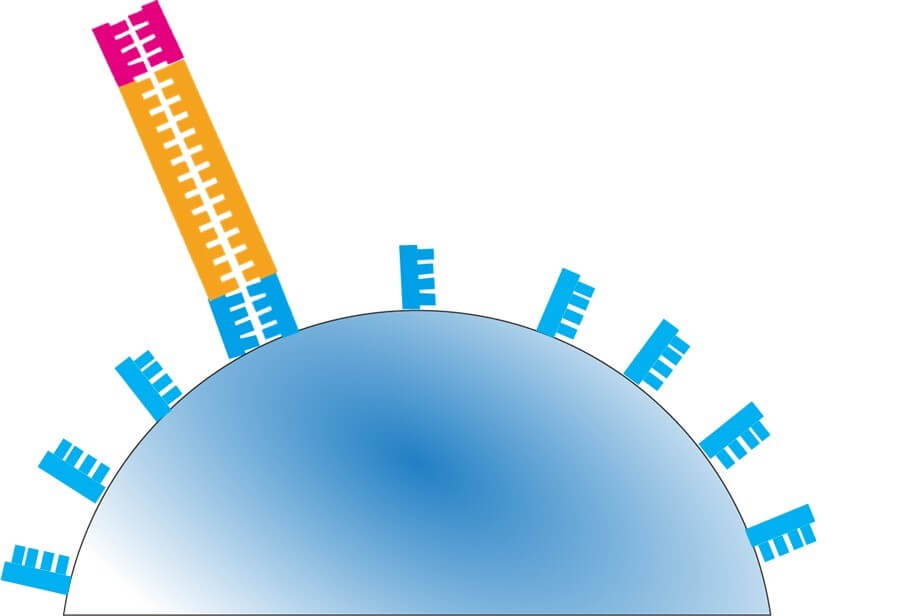
解離したDNA断片はビーズ上の近隣の断片に再び接着する。
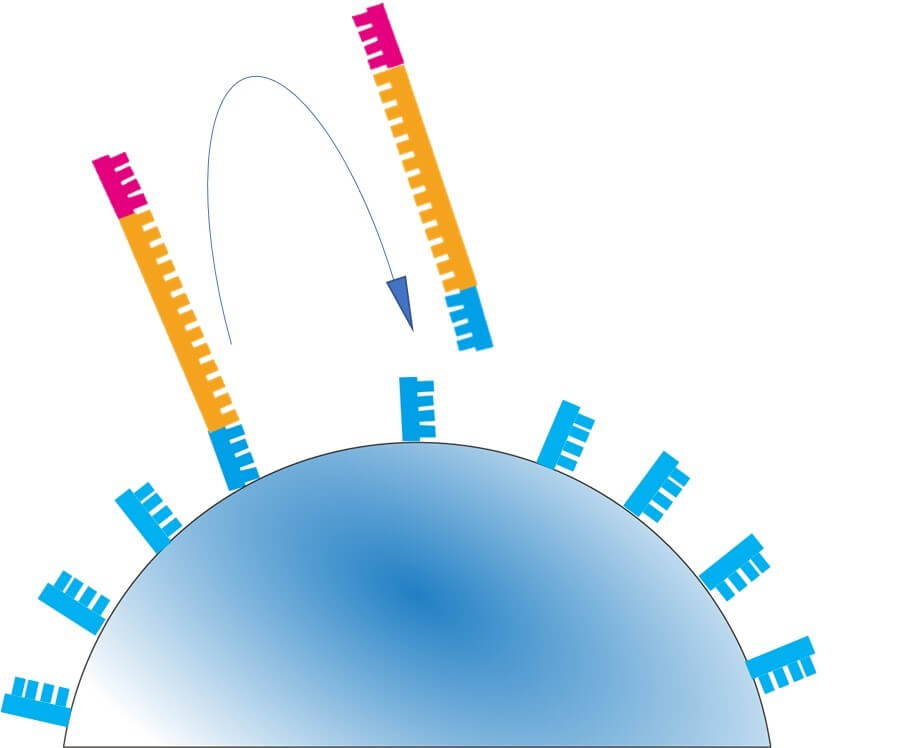
接着した遺伝子断片を鋳型に伸長が行われる。
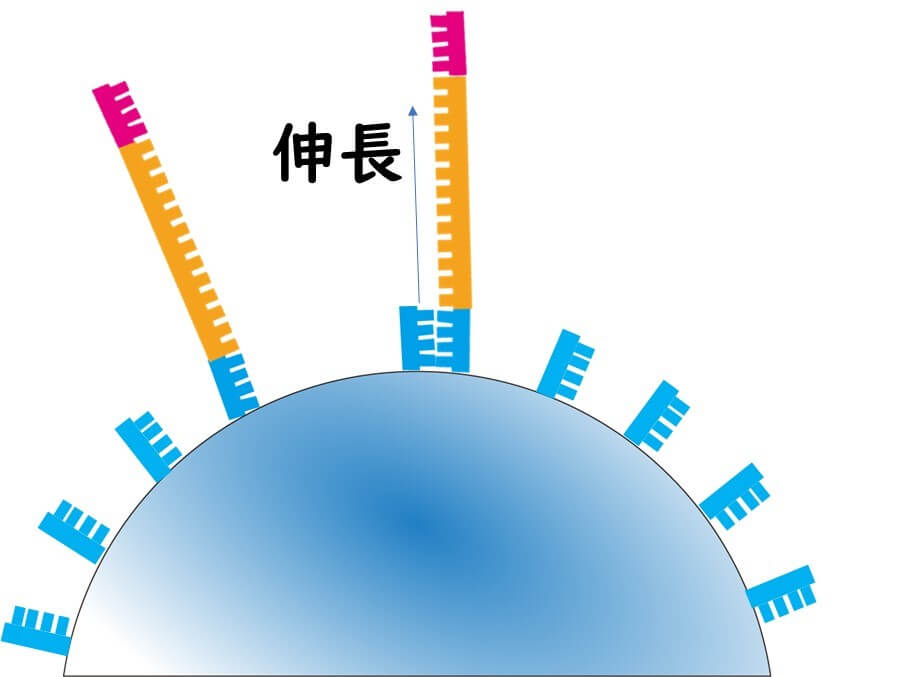
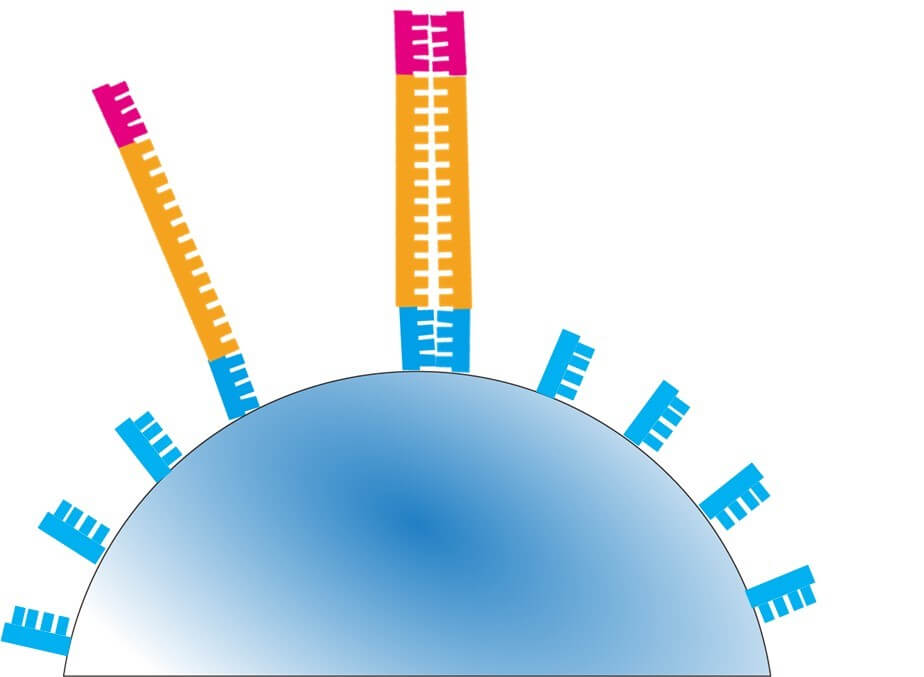
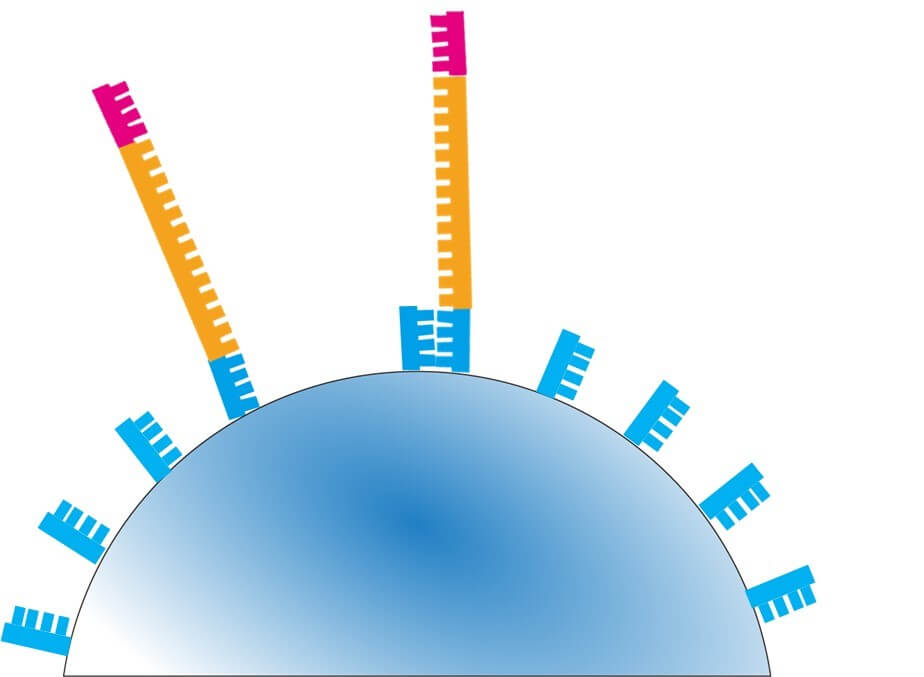
伸長の鋳型に用いられたDNA断片は再び熱変性により解離する。そして、 解離したDNA断片はビーズ上の近隣の断片に再び接着する。
以上の過程がビーズ表面で繰り返される。
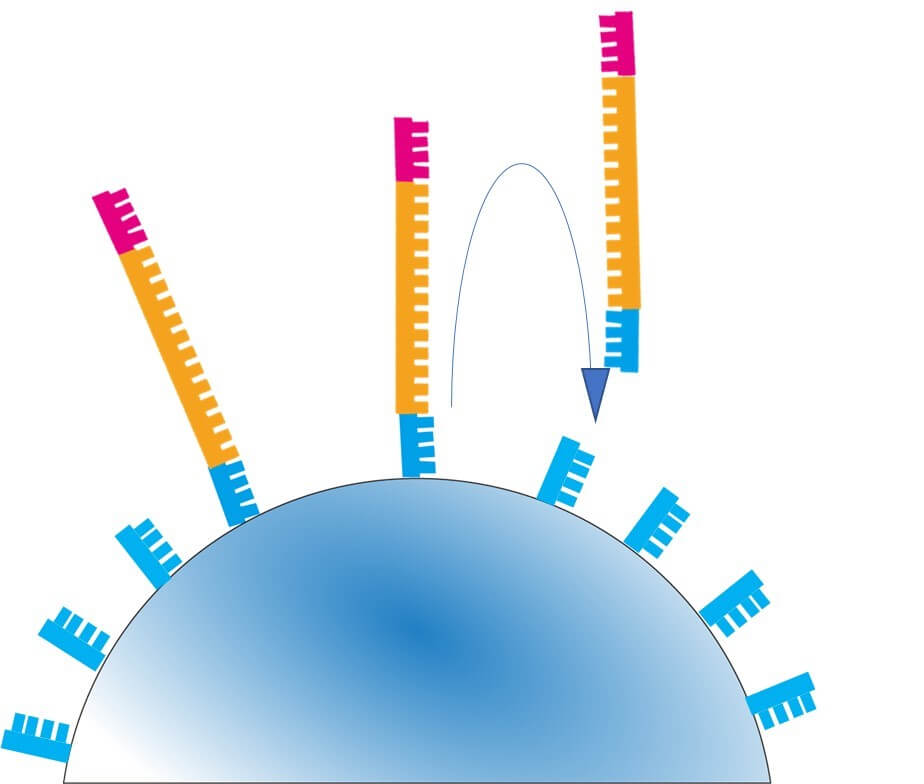
エマルジョンPCR増幅後では、1つのビーズの上に1種類の目的DNA断片のみが増幅されている。上述したように、ビーズと目的DNA断片とのモル比をあらかじめうまく調整してあるので、多くの場合1ビーズには1種類の目的DNA断片のみが増幅されていることとなる。そうでないビーズにに関しては、DNA配列解析する段階でノイズとして検出され、廃棄されるデータとなる。
このビーズを 専用の半導体チップ上に並んだ数μmのウェル(穴) に入れる。 半導体チップ上 には約 1,200 万ウェルがある。以下の反応はこれらのウェル内で同時に進行する。
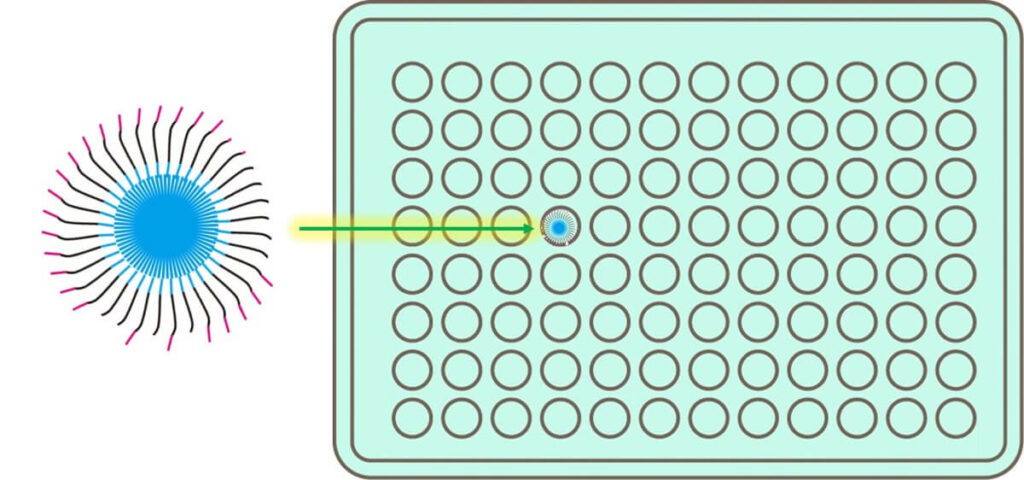
Ion Torrent方式の最も大きな特徴は、その塩基配列の決定方法である。他の多くのシークエンサーは異なり、1塩基の増幅ごとに放出されるH+によってもたらされるpHの変化を測定するというものだ。エマルジョンPCRによって増幅されたマイクロビーズを半導体チップの小さなホールに一つ一つ一つ埋め込む。そしてその一つ一つのホールにおいてこのようなpHの測定が行われる。このように半導体を塩基配列の測定に用いているという手も大きな特徴である。
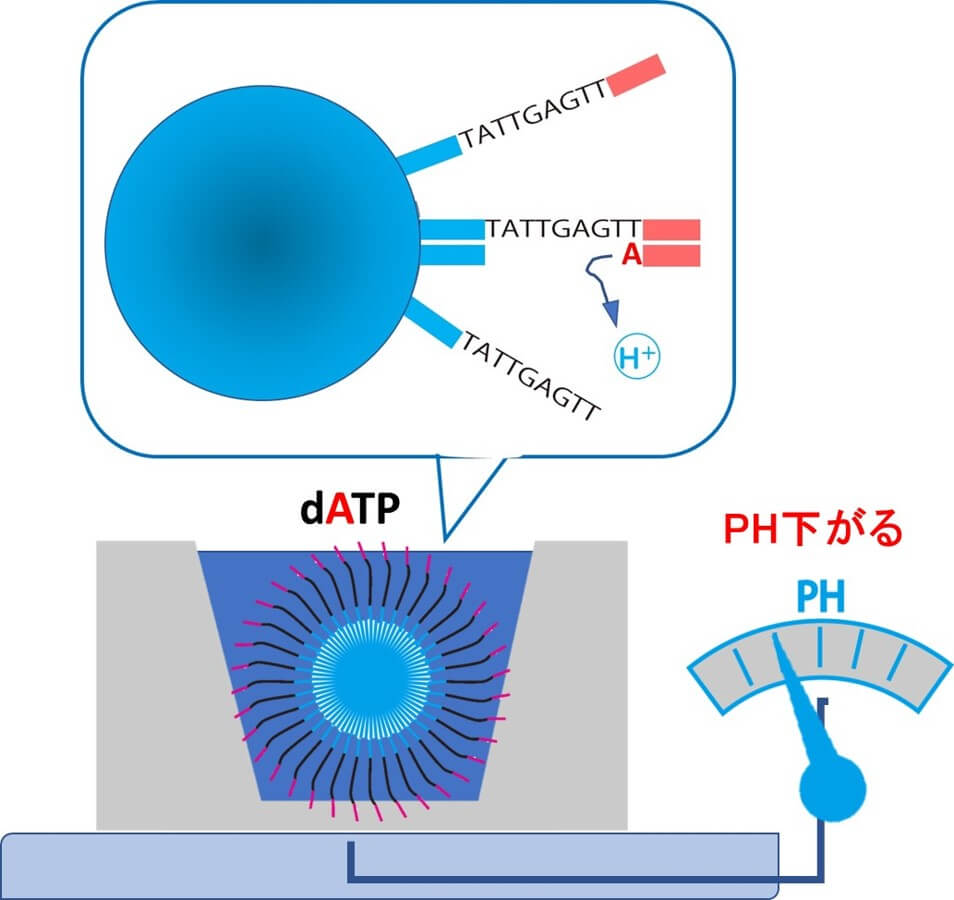
1)ヌクレオチドのAの反応液でウェル内を満たす。
2)鋳型DNAの塩基はTなので、この場合ポリメラーゼの伸長反応が行われる。
3)この塩基の伸長が行われる際、 H+にが放出される
4)結果として、ウェル内のPHが下がる
5)このPHの低下を高感度測定する
すなわち、あるヌクレオチド液を与えたときにpHが下がれば、そのヌクレオチドが「当たり!」のヌクレオチドということになる。
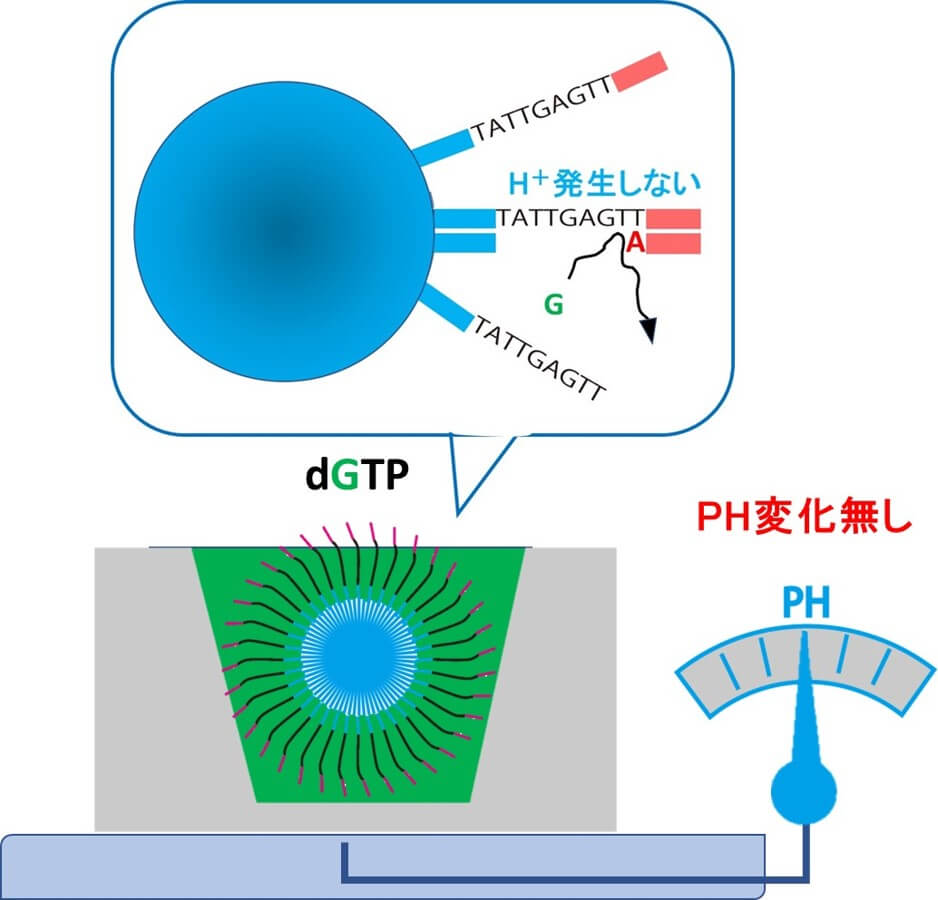
1)つぎにGのヌクレオチド液でウェル内を満たす。
2)この場合、鋳型DNAの次の配列はTなので、相補的なヌクレオチドはAである。Gのヌクレオチド液でウェル内を満たしても、塩基の伸長は行われない。
3)すなわち、 H+の放出はおきない
4)ウェルのPHは低下しない。
すなわち、あるヌクレオチド液を与えたときにpHが下がらなければ、そのヌクレオチドが「はずれ」のヌクレオチドということになる。
以上のようなプロセスが半導体チップ型のマイクロチップ上の 約 1,200 万ウェルで同時に進行する。
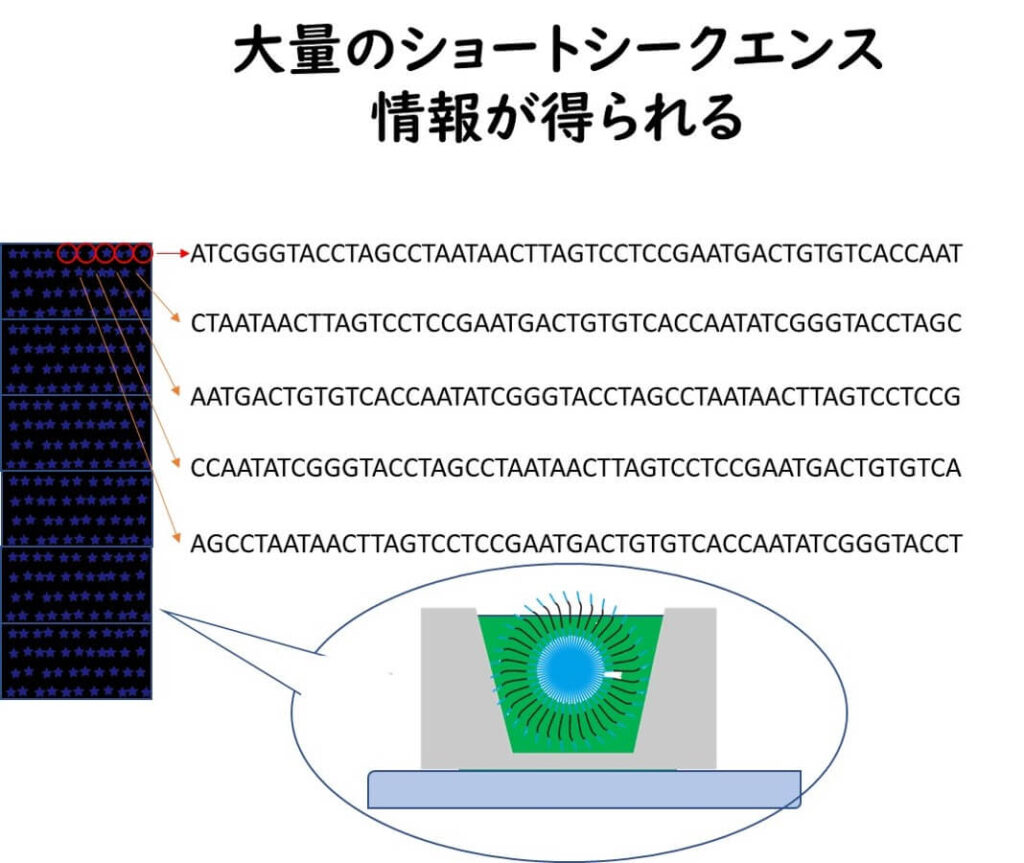
このようにして塩基配列が決定された無数のショートシークエンスをコンピューター内でつなぎ合わせる作業を行う。
うう
以上のプロセスを下記動画にしてみました。
食品微生物検査と次世代シークエンサー
次世代シーケンサーが登場してから15年余りが経過した。次世代シークエンスを用いることにより、これまでのサンガー法に比べて、桁違いにハイスピードで、また低コストで、DNAの配列を読むことができるようになった。食品の微生物検査において 次世代シーケンサー のもたらす技術は、
1)コロニーから細菌のすべてのゲノム配列を決定する方法と
2)食品から直接複数の微生物のゲノムを網羅的に抽出する方法
の2つに大別することができる。どちらの技術もこれまでと異なる景色を食品の微生物検査の世界ににもたらしていくだろう。
過去10年間、特に直近の過去5年間の研究の進展を見ると、コロニーからのゲノム配列の研究の進展が著しい。特に食中毒細菌の分子疫学分野における応用が著しいスピードで行われている。その中でcore genome multilocus sequence typing(cgMLST)の登場がもたらすインパクトは大きい。cgMLSTにより得られた情報は、世界のどこででも誰もが共通の言語として同じ尺度で使用できるからだ。このような技術は、公共機関が行う食中毒の分子疫学解析のへ応用にとどまるだけではなく、食品企業などが行う日常の微生物検査にも広く応用できるだろう。
※ cgMLST など次世代シーケンサーを用いた食中毒菌の分子疫学解析の基礎については下記の別記事でわかりやすく解説していますので、ご覧ください。
病原菌や食中菌の感染ルート把握のための分子疫学解析手法(菌株の識別法)のすべてをわかりやすく解説します
一方、食品から直接多様な微生物のゲノムを抽出する方法、特に16SrRNAアンプリコンシーケンスにより、培養法に比べると格段のスピードで細菌叢を解析できることが可能となった。今後、特に食品の品質管理や原料管理などの分野において、大きな可能性を秘めている。
第3世代シークエンサー法(ナノポアシーケンシング)
さらに、次世代シーケンサーのショートリード(300bp程度の短断片しか読めない)という欠点を補うロングリード(100Kbpを超える塩基配列を一度に決定できる) の第 3 世代シークエンシング(Third Generation Sequencing、TGS)も登場した。これらの新型シークケンス技術は実用的には2017年ころから急速に世界で普及している。
第三世代シークエンサーの代表的な技術は、Pacific Biosciences(PacBio)とOxford Nanopore Technologies(ONT、ナノポアシーケンス)であり、どちらもNGSに比べるとロングリードであることが特徴である。特にナノポアシーケンスは、ヌクレオチドの合成を伴わない。本技術は、2014年にOxford Nanopore Technologies(ONT)よりMinIONプラットフォームとして発売された。
本記事では、ナノポア―シークエンスを例に挙げ、解説することとする。
原理
ナノポアシークエンス法では、DNA分子やRNA分子を、あらかじめポリマー製の膜に埋め込まれたタンパク質であるナノポアに通過させる。このポアの直径が数ナノメートル以下であることからナノポアと呼ばれる。ポリマー製の膜には通電性があり、この膜に電圧をかけることで、ナノポアにイオン電流を流すことができる。そして、このナノポアにDNAを通すと、通過するヌクレオチド毎に特徴的な電流の乱れが発生する。このように、異なるヌクレオチド塩基は、ナノポアを通過する際の電流の変化によって区別される 。
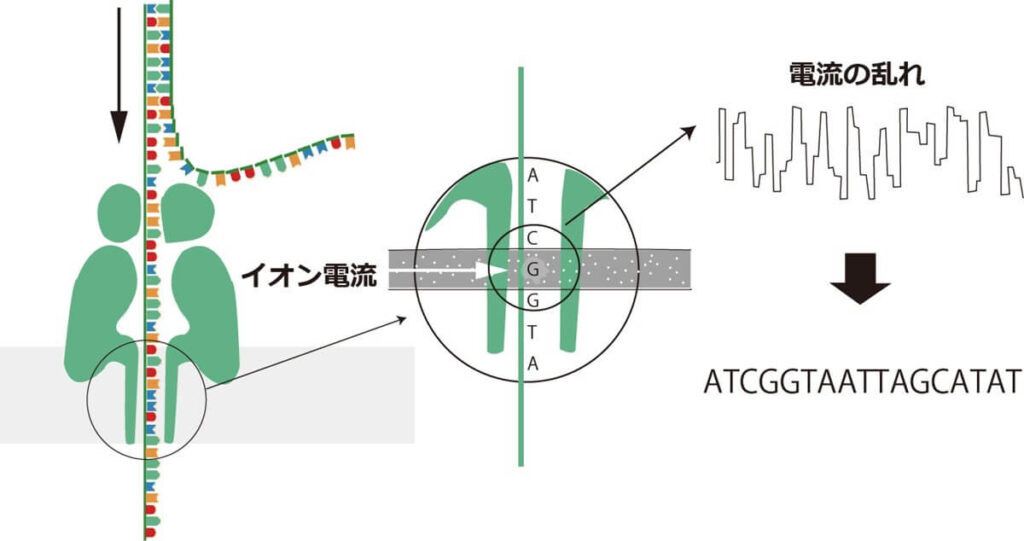
ナノポアシークエンスが、これまでのシークエンス技術と根本的に異なる点は、塩基配列決定のプロセスでヌクレオチドの合成を行わない点にある。サンガー法や次世代シークエンサーのすべて、また、第3世代に分類されるPacBioのいずれもが、ヌクレオチドの合成反応を伴う。しかしナノポアシーケンスでは、ヌクレオチドの合成反応を一切行わない。これが低ランニングコストと操作の簡便性をもたらしている理由だ。
なお、原理の動画については、下記のOxford Nanopore Technologies社の解説サイトで大変わかりやすい動画が公開されいる。https://nanoporetech.com/applications/dna-nanopore-sequencing
食品微生物の検査において次世代シークエンサーに比べての利点
食品微生物の検査において ナノポアシーケンシングが次世代シーケンシングに比べて優れている点は下記のとおりであある。
1.ロングリード
第1に、次世代シークエンサーシーケンサーのようにショートリードではなく、長いリードレングス(10,000-50,000bp以上)が可能である。上述したように塩基配列決定のプロセスでヌクレオチドの合成を行わず、ナノポアに測定対象のDNAを通していくだけである。したがって、理論上では無限の鎖長の塩基配列を読める(ただし、実際はサンプル調製の関係で平均10k塩基長となっている)。ショートリードの次世代シークエンサーシーケンサーは、反復配列を多数含む塩基配列決定が苦手であった。短い断片の張り合わせる際、繰り返し配列が多く含まれると、接続領域がわからなくなるからだ。これに対して、ナノポアシークエンスでは、一本のDNA鎖を連続的に読むので、このような繰り返し配列領域のシークエンスを間違いなく行うことが出来る。また、後述するようにメタゲノム解析において1細胞由来のコンプリートゲノムの抽出を行うことも可能である。
次世代シーケンサーは、食品微生物検査においては、現時点では、 16SrRNAアンプリコンシーケンス などの技術により食品中の微生物菌叢を調べることなどに活発に用いられている。ただし、次世代シーケンサーは300bp程度の短い一部の16SrDNA断片の情報だけで菌叢を推測している。したがって、その精度は属れべるにとどます。一方、第3世代シーケンサーでは16SrDNAの全長解析が可能なので、より精度の高い種レベルの菌叢解析が可能となる。
2.ヌクレオチドの合成反応不要(簡便操作)
第2に、ヌクレオチドの合成反応を伴わないので、NGSより短時間で準備が可能である。非増幅のゲノムDNAを使用するため、酵素、クローニング、増幅のステップが不要である。従って、分析のランニングコストも安い。また、分析時間も短い。このようにサンプル調整がNGSより簡便なため、直接現場でシーケエンスを行うリアルタイム分析も可能である。
このようなナノポアシーケンシングの利点を生かして、手のひらサイズのポータブル型のシークエンサー「MinION」も開発されている。 MinION は多くの異なる環境で実験室の外のさまざまな現場で使用が期待されている。
食品微生物の検査では、たとえば、工場でのリステリア菌やサルモネラ菌の汚染がある場合に、工場サイトレベルで簡便にこれらの菌の検出・同定に応用できる可能性が期待されている。

食品微生物の検査に応用するための技術的課題
ナノポアシークエンスの現時点での最大の欠点は、シーケンスエラー率が高いことである。これまで使用されてきた天然または人工のナノポア構造の中には、DNAがポアを通過する際に、一度に1つのヌクレオチドだけの特徴を検出するのに適した形状のものはなかった。これらのナノポアには5nm以下のチャネルはなく、少なくとも10~15ヌクレオチドのssDNAがこの長さのチャネルを通過するため、これらのヌクレオチドすべてがイオン電流の遮断に寄与する。これがシークエンスエラーの原因となっている。
現段階の技術では、DNAの移動を1塩基あたりのマイクロ秒からミリ秒に遅らせることが短期的な課題となっており、装置の改良が続けられている。最新のバージョンでは新しいタイプのナノポアを利用しており、最大99.99%のシークエンス精度を提供できるとメーカー側が情報提供を行っており、その精度は急速に向上中である。食品微生物の検査の実用に応用できるレベルに急速に近づいている。