病原菌や食中毒菌の感染ルートの把握には、精度の高い鑑別法(fingerprinting)が求められる。これまでに細菌の株レベルのタイピングは主としてパルスフィールドゲル電気泳動法に代表されるフラグメント解析や、キャピラリーシーケエンサー(サンガー法)により一部の遺伝子配列を読むMLSTやMLVA等が主流であった。しかし、今後、細菌の株レベル分子疫学解析は、次世代シークエンサーによる全ゲノム配列解析が主流になる。本記事では、これらの分子疫学解析法(菌株の識別法)のすべてを、わかりやすく説明する。
株識別の重要性
まず、細菌の株識別の重要性について説明する。
食中毒事件が起きた際に、公的衛生機関がその原因菌を推定するための分子疫学的解析を行うためには、 16 S rRNA遺伝子(16SrDNA) による種の同定だけでは不十分である。
例えば、下の図で、これが狐なのか猿なのかこれホモサピエンスなのかということについては 16SrDNA でも分かる。このような種のレベルの判別は 16SrDNA によって可能だ。
※遺伝子解析による細菌の種の同定法の基礎を知りたい方は下記の記事もご覧ください
遺伝子検査による微生物の同定

今、例えば、地球を遊覧飛行中の宇宙人船が、地球上の人類の何者かに撃墜されたとしよう。
宇宙人の隊長は部下の宇宙船を撃墜した犯人は誰かを探す。

しかし、この際、その犯人がホモサピエンスであることしか知りえないなら、宇宙人は地球上の全人類を滅ぼさなくてはならない。しかし実際にUFO円盤を撃墜したのは、地球上のホモサピエンス種のある特定の人間である。このように人間一人一人の顔を識別するする方法を細菌学では分子疫学解析(菌株識別)と呼ぶ。
分子疫学解析 手法を使えば、宇宙人はこの特定の人間だけを逮捕すればいいということになる。 病原菌や食中毒菌の感染ルートの把握も、これと同じだ。分子疫学解析 手法は、ある細菌種のなかの特定の株のみを識別する方法である。

細菌の株の識別タイピングには 全ゲノム情報が必要
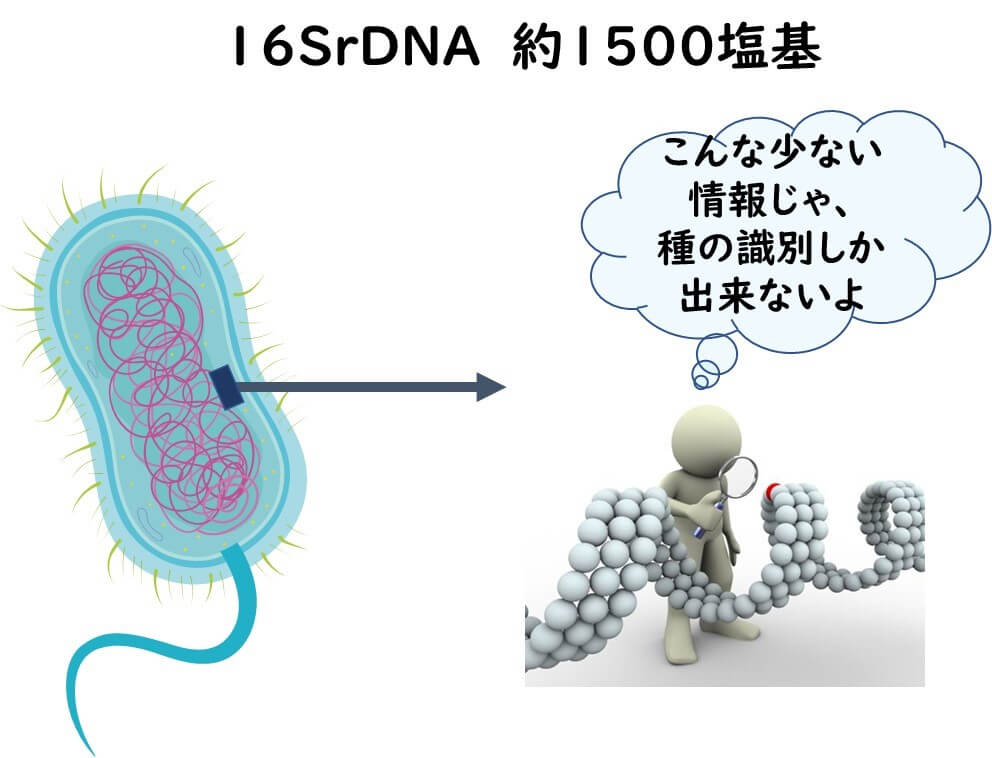
さて、細菌の株の識別タイピングは、種の同定とは異なり、1500塩基程度のDNAから抽出される遺伝的情報では、解像度の点から、十分な識別の目的を達することはできない。
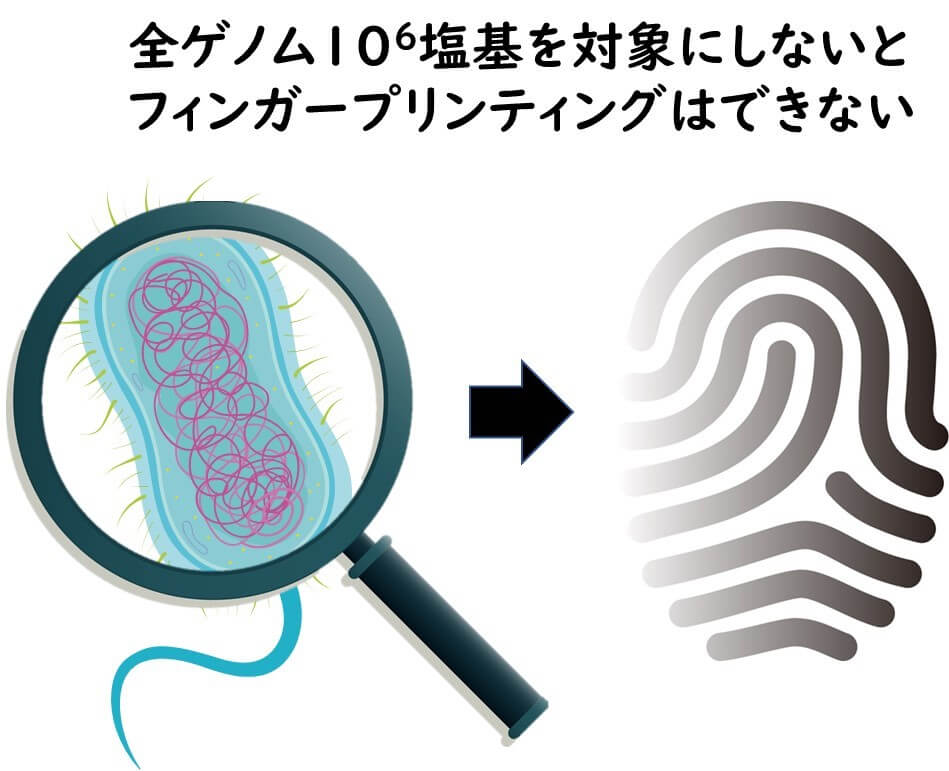
細菌の持っている全ゲノムは約106bpである。株レベルの識別には全ゲノムから抽出される遺伝情報が必要となる。
パルスフィールドゲル電気泳動(pulsed-field gel electrophoresis: PFGE)
1990年代に登場した パルスフィールドゲル電気泳動(pulsed-field gel electrophoresis: PFGE)は2010年ころまでは、世界の細菌の分子疫学解析の主役であった。
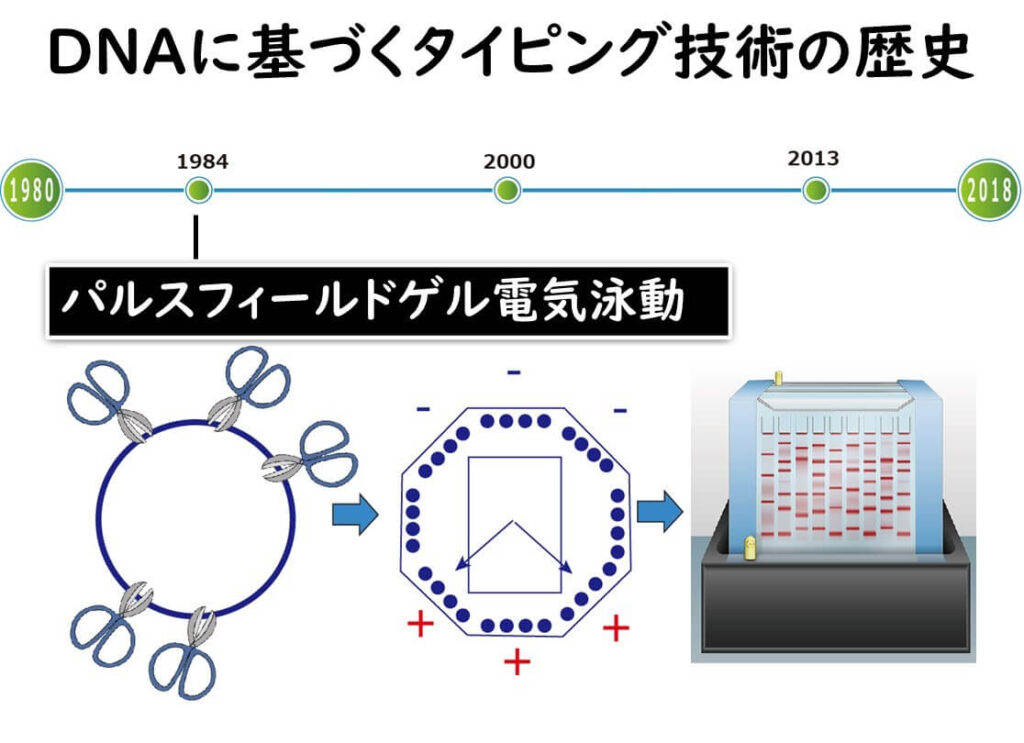
パルスフィールドゲル電気泳動 はフラグメント解析である。フラグメント解析とは、ゲノムを制限酵素で切断する、あるいは、ゲノム中の特定の領域をPCR法などで増幅することにより、一定の長さのDNA断片の出現パターンを電気泳動上で比較する技術である。本来、細菌を株レベルで識別するには、細菌のもっている全ゲノムからの情報の比較が基本となる。しかし、 パルスフィールドゲル電気泳動 が登場した1984年当時、全ゲノムの配列を読むことはおろか、特定の一部の遺伝子配列を読むことすら、技術的に困難だった。そこで、 パルスフィールドゲル電気泳動 は1990年代以降現在に至るまで、現場レベルでこの目的を達することのできる数少ない方法の一つとして普及してきた。現在でも、多くの国の分子疫学解析では、 パルスフィールドゲル電気泳動 が用いられている(後述するように2019年米国は使用を中止した)。
パルスフィールドゲル電気泳動 は細菌のゲノムを制限酵素(遺伝子の特別な配列だけを認識してその部位で切断する酵素)処理をして、切断された断片の長さの違いを解析する手法である。
例えば16SrDNAなどのように1500bp程度の短い断片を制限酵素で切断した場合にはひとつの遺伝子断片が100から300 bp程度になるので、直流電極を用いた電気泳動で電気泳動をすることができる。一方向のみの電極で引っ張った場合には DNA はひも状に直線となり、プラス電極方向へ引っ張られていく。小さな DNA 断片の場合にはアガロースゲルの中をこのようなやり方でも通過することができる。しかし大きな DNA 断片となった場合には DNA が伸びたままでアガロースゲルの中を移動できない。
※電気泳動の基本事項の確認については下記の記事の電気泳動の部分をご覧ください
PCR法とリアルタイムPCRをわかりやすく説明します
パルスフィールドゲル電気泳動では106の全ゲノムを制限酵素で切断する。したがって切断された断片の大きさは、少なくとも104 bp程度の大きな断片となる。このような大きな遺伝子断片は単純に直流電極で一方向に引っ張っても移動することができない。そこでパルスフィールドというプラスとマイナスの電極の方向をパルス時間と呼ばれる時間間隔で交互に切り替えるわけだ。電極の方向を変えることによって、紐状に伸びたDNAはいったん収縮し、 ランダムコイル状に戻る。 そして新しい電極の方向に向かって進むと考えられている。
原理のこれ以上詳細な説明はここでは割愛するが、要するに「全ゲノムを制限酵素で切断した大型DNA断片は、電極の方向をちょこちょこと切り替えることによってはじめて、だましだまし移動させることができる」考えれば良い。
パルスフィールドゲル電気泳動法では、異なる分子量の断片の数の違いにより株の分別することができる。 パルスフィールド電気泳動法 では全ゲノムを対象とした解析手法なので、16S rDNAやその他のタンパク質コード遺伝子の全長の塩基配列(通常、1000~2000塩基)を読むよりははるかに高い解像度が得られる。
パルスフィールド電気泳動法 は病原菌や食中毒菌の疫学解析に広く用いられており、1996年には、PulseNetと呼ばれる国際ネットワークもCDCを中心に設立されている。これは パルスフィールドゲル電気泳動法 によるタイピング法を標準化し、得られた遺伝子パターンをデーターベース化するもので、ネットワークにアクセスする研究室では食中毒発生時に、CDCのデーターベースと照合を行えるようになっている。
フラグメント解析の限界
パルスフィールドゲル電気泳動法 の技術的特徴は、フラグメント解析技術であるという点である。 フラグメント解析 はいくつかの欠点を持っている。
- 識別精度とデータ互換性
フラグメント解析技術は、すべて、原則として電気泳動ゲル上での遺伝子断片の数や移動度を解析するものである。このようなフラグメント解析の最大の欠点は、解析するフラグメントの採否をめぐるバンドの濃さの閾値や移動度の微細な差異に関する判断などについて実験者や研究者毎の違いが生じる可能性があることである。また、そもそもこの手法は遺伝子の断片の分子サイズを見ているに過ぎないという欠点がある。同じ断片のサイズであっても配列が異なる可能性もある。
また、国際的に複数の研究室でのデータの互換性が乏しい。 パルスフィールドゲル電気泳動 法における国際的な共有データベースのPulseNetもその例外ではなく、実験者や研究室での解析技術によるフラグメントデータの標準化を達成するためには煩雑な手間を要する。つまり、フラグメント解析は、本質的には系統解析(菌株間の進化的距離の解析)には向いていないと技術といえる。

- 簡便性
また、 パルスフィールドゲル電気泳動法 の欠点は、実験操作が煩雑であり、解析に長時間を要するという点である。 パルスフィールド電気泳動法 のこのような欠点を補うために、特に食品の民間企業では、過去20年間、AFLP法やRibotyping法などのフラグメント解析も病原菌や食中毒菌の株の識別に用いられる場合があった。ここでは、紙面の都合上、これらの手法の説明は省略するが、Ribotyping法については、リボプリンターなどの全自動装置も販売されており、簡便性という点では優れているが、装置やランニングコストも高いという欠点がある。また、株識別の解像度はPFGE法より劣る。また、AFLP法も、操作は決して簡便とはいえなかった。
要するに、これまでフラグメント解析が広く用いられてきた理由は、「配列を読む技術がない」「配列を読むと時間がかかる」「配列を読むとコストがかかる」といった技術的、経済的理由からである。歴史的にみると、たとえば、1990年代初頭、PCR法が生物学の領域に登場し、生物のDNA配列が研究者にとて身近になった頃、細菌学の分野では種の同定を16S rDNA配列で決定する技術が導入された。しかし、当時は、DNAシークエンサーは高価な装置であり、大学や研究機関に1装置導入するのがやっとの時代であったため、手元の細菌の16S rDNA配列を簡単に読むことはできなかった(わずか、1500塩基であっても!)。そこで、当時もっぱら、行われたことは、16S rDNA増幅断片の全塩基配列を読むかわりに、これらを制限酵素で切断し、出現するフラグメントパターンでおよその細菌の同定やグルーピングを行う方法だった。これは、RFLP(Restriction Fragment Length Polymorphism、制限酵素断片長多型)とよばれ、1990年代には 16S rDNA の配列を読む代わりに盛んにおこなわれた。16S rDNAにかぎらす、当時は、1500塩基程度の塩基配列を読むことも、時間的、経済的に、研究者にとって大変敷居が高く、上記のようなフラグメント解析が盛んに行われた。しかし、このような1遺伝子レベルのフラグメント解析は2000年頃になると、キャピラリーシークエンサーが登場し、DNA配列解析技術の迅速化、低コスト化にともない、ほとんど行われなくなった。
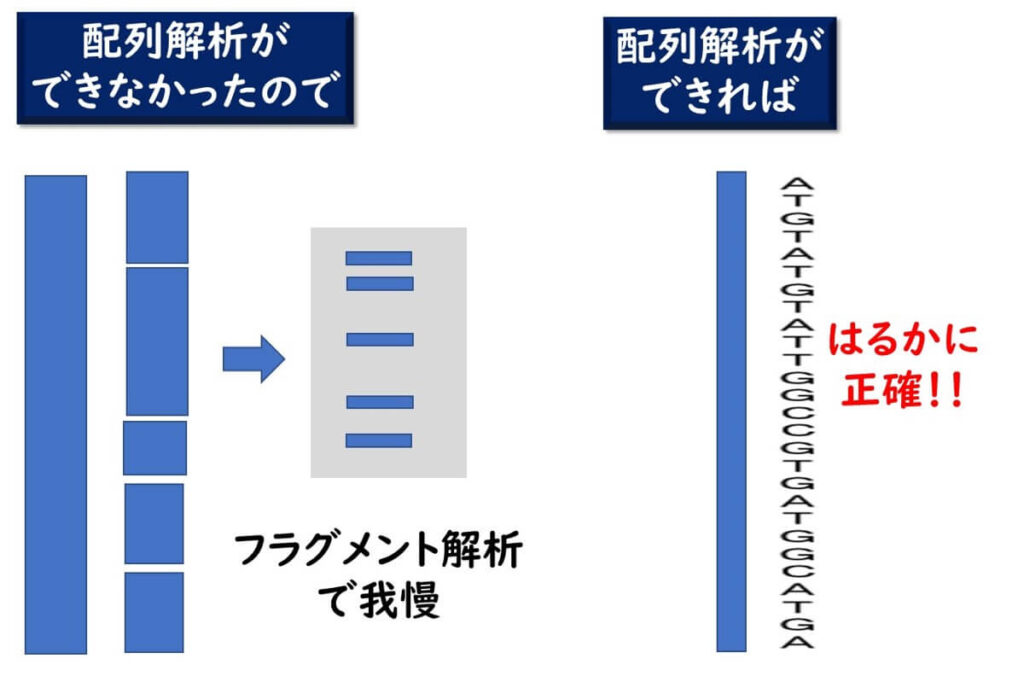
そして、2010年以降、次世代シークエンサーが登場した。細菌の全ゲノム解析が低コストで迅速に行われるようになって10年経過している。もはや全ゲノムを対象にしても、ゲノムのシーケンスをすることは、時間的にもコスト的にも難しくはなくなってきた。したがって、 パルスフィールドゲル電気泳動 も、2000年以降に16 S DNA のRFLPが辿った歴史(消滅)と同じような歴史ををたどると予想される。すでに米国では、2019年、CDCが用いる分子疫学解析手法を パルスフィールドゲル電気泳動 を廃止した。そして、全面的に次世代シークエンサー に基づく全ゲノム配列解析に移行している。
DNAシークエンスに基づく分子疫学手法
2000年になり、キャピラリー方シークエンサーが急速に普及した。このようなDNA解析の高速化、低コスト化潮流の中で、細菌のDNA解析においても多数の菌株についてそれぞれ5~6遺伝子領域を同時に数百塩基ずつを日常的に解析することが可能となった。そこで、登場してきたのが Multilocus sequence typing(MLST)法 である。MLST は、2000年に登場後、2022年の現在に至るまで、多くの食中毒菌や病原菌で活用されている。
Multilocus sequence typing(MLST)法
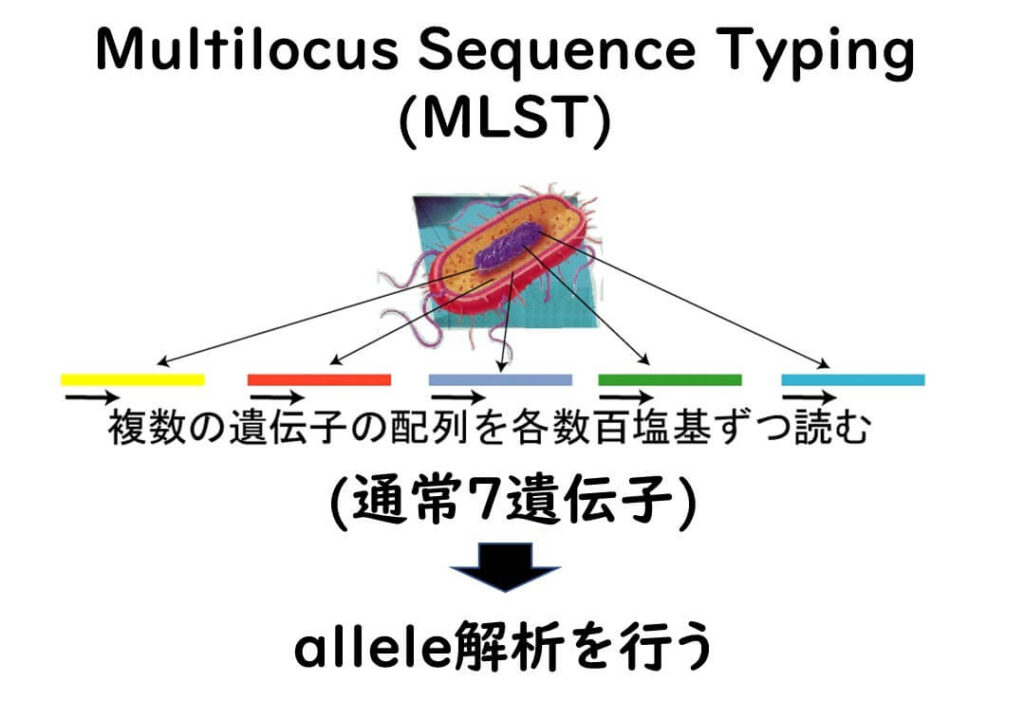
MLST法では微生物の全ゲノムの中から7つの遺伝子を選び、その配列を読む。この手法が登場した2000年当時は、微生物の7つの遺伝子の部分配列を読むぐらいなら、公的機関の1研究室でも十分可能になっていた。

本手法は、複数の遺伝子領域(通常7領域以上)のそれぞれ400塩基程度の配列を読み、それらをもとに菌のタイピングを行う方法である。菌株毎に複数遺伝子の配列の差異をパターン化して(allelesに分別)、それらを統合遺伝子解析ソフトにより総合的に解析することにより、 パルスフィールドゲル電気泳動 法以上の解像度での株識能を達成することが可能となる。
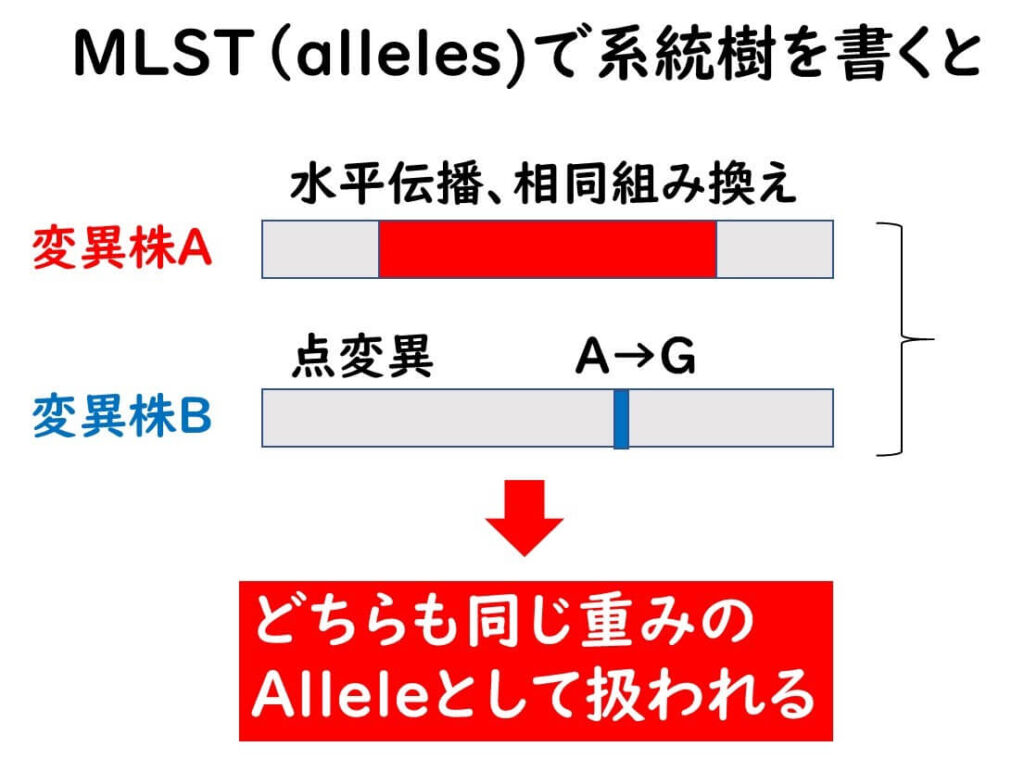
これまでの多くの遺伝子解析による系統樹の作成の仕方と異なるのは、各遺伝子の配列そのものから系統樹を書くのではなく、各遺伝子の配列の差異をグループ化して、そのグループ番号から系統樹を書くものである。つまり、ある遺伝子で、A株、B株、C株の間に遺伝子配列の違いがあればすべて異なるグループ(alleles)に分類される。

しかし、これまでの遺伝子配列をもとにした系統樹作成法と異なり、A株とB株との間の遺伝子配列の異なる塩基数は問題とならない。例えば、A株B株の間に1塩基のみの差異があり、A株とC株の間に10塩基に差異があった場合、これまでの遺伝子配列による系統樹ではA,B株は近く、C株は遠い位置に系統樹が作成された。しかし、MLSTではA,B,C株の差異は均等に扱われる。この理由は後述する。

MLSTの利点
MLSTの最大の利点は遺伝子配列情報に基づくため、結果がデジタルであることが挙げられる。 パルスフィールドゲル電気泳動 法は最終的に人間の視覚によってバンド位置を検出する(アナログである)ため、主観が入ってしまう可能性があった。
MLST解析は遺伝子配列情報に基づくものであるので、「曖昧さ」がなく、どの研究室で行ってもデータの比較がしやすいのは大きな利点である。そのため、文字列としてデータベースに登録しやすく、複数の研究機関で株データを共有するのに適している。本来、世界で共通のデータを共有しようとするなら、遺伝子配列情報に勝るものはない。ATGGT・・といった配列情報は、世界のどの研究室で解析しても同じであり、正確で共通なデータがインターネット上で交換できる。もちろん、これまでも、ある特定の遺伝子配列(16S rDNA、gyrBなど)による微生物の種レベルの識別は行われてきた。しかし、株レベルの識別には1つや2つの遺伝子(通常1000塩基程度)の配列情報の解像度では、とてもその目的を達成することができない。このため、遺伝子配列情報で株の識別をすることはこれまで行われてきていなかった。
また、MLST法は パルスフィールドゲル電気泳動 法法と異なり、操作は遺伝子配列を決定するとういう単純な操作で実施できる。その後の配列編集や系統解析なども、便利なソフトが市販されているので、難しい作業ではない。また、マルチキャピラリーシークエンサーを用いれば、操作のかなりの部分が自動化されている。
なぜ複数遺伝子配列(Multi Locus)なのか?
ここでMLST法が登場してきた理論的背景について改めて解説しておく。前述したようにMLST法登場は、DNA解析の高速化、低コスト化への潮流の中で自然の成り行きとして登場してきた。
しかし、もう一つの理由として、細菌の系統に対する私たちの認識が過去20年間での大きく変化した点もあげられる。過去20年間で、細菌のゲノム進化に対するわれわれの認識は、細菌集団をclonal populationと捉えることから、non clonal populationと捉える方向に変化した。1990年代、私たち研究者の間で自分の扱う細菌の遺伝子配列(多くの場合16S rDNA)を読むことができるようになった時点では、細菌遺伝子の進化は主に点変異によっておき、それが子孫に伝わっていくという考え方が主流であった。ところが、2000年以降、世界で種々の細菌のさまざまな遺伝子配列が明らかにされると、細菌遺伝子の進化は必ずしも点変異だけではなく、水平伝播になどよる大きな遺伝子断片の相同組み換えが私たちの想像していた以上に頻繁におきていることが次第に明らかとなってきた。
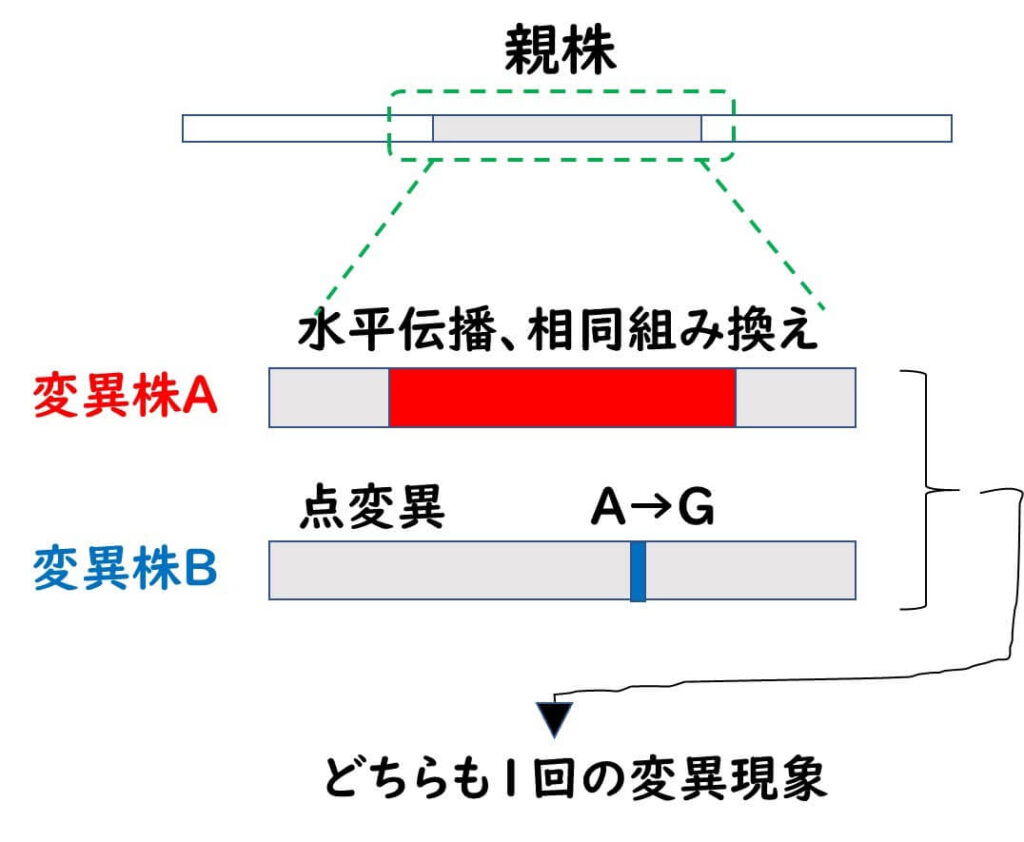
細菌遺伝子の進化が、点変異の蓄積のみが子孫に伝わっていく結果と考える場合、私たちはその細菌集団をclonal poplulationと呼ぶ。一方、相同組み換えなど大きな遺伝子断片の交換が頻繁に起きていると考えられる集団をnonclonal populationと呼ぶ。Clonal populationであったなら、その細菌の系統はたった一つの遺伝子の配列からつくられた系統樹でもほぼ正しく系統を反映することができる。しかし、nonclonoal population の場合、ひとつの遺伝子だけで系統樹を書くと、仮にその遺伝子内で一回大きな遺伝子断片の相同組み換えがおきていたとすると、その組み換えをおこした株のみは、他の株に比べて系統的に遠くかけ離れたところに置かれてしまう。一方、大きな断片の相同組み換えを起こしていないものの、点変異を複数回おこした株は、本来、上記A株より系統的(つまり進化時間的に)にはるかにかけ離れている存在であるが、上記A株より元株に近い位置に系統樹が書かれてしまう。
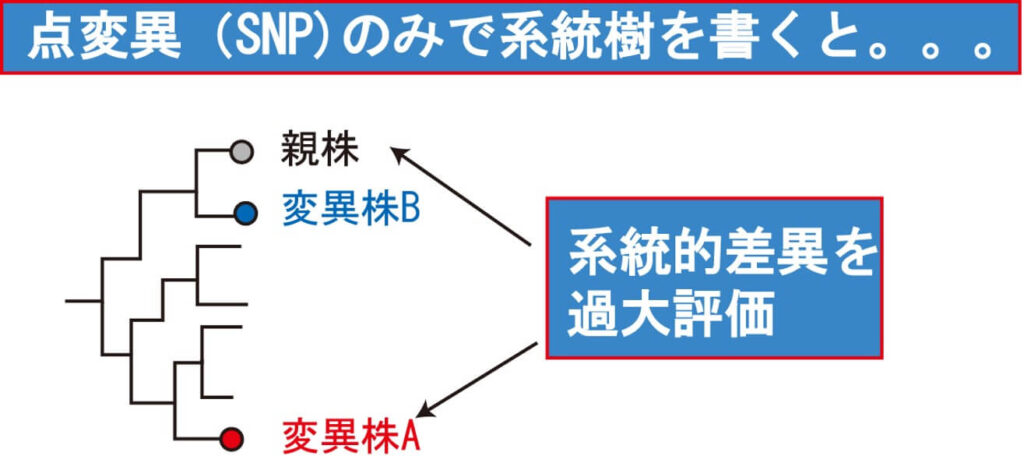
このように、相同組み換えが頻繁に起きるnonclonal popultionの場合、たった一つの遺伝子で系統樹を書くのは系統を誤る可能性が高い。MLSTでは、
1) 複数の遺伝子配列情報を元に系統を書く
2) 遺伝子配列そのものを元に系統をかくのではなく、遺伝子配列の差異(alleles)にもとづいて系統樹を書く
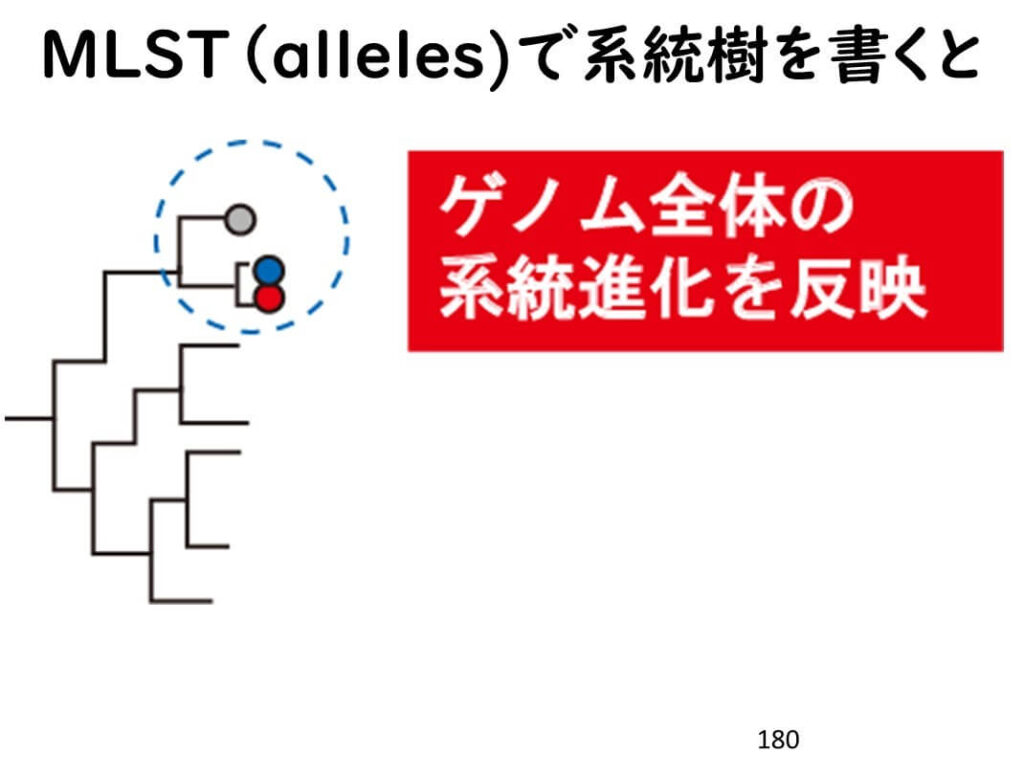
このため、nonclonal populationについても実際におきた進化的時間軸を正しく反映した系統樹がかけるわけである。
MLST解析に用いる遺伝子は?
次に、MLST解析に用いるべき遺伝子について述べることとする。細菌のタイピングに用いる遺伝子を大別すると、 ハウスキーピング遺伝子 とその他の変化しやすい遺伝子に2分できる。ハウスキーピング遺伝子とは、細菌の生存の根幹にかかわる遺伝子であり、たとえばリボゾームの遺伝子(16S rDNA)やDNAの複製に関与するジャイレース遺伝子(gyrB)、ヒートショックタンパクなどのストレスタンパク遺伝子などがこれにあたる。 ハウスキーピング遺伝子 の変異は細胞にとって根幹的な作用に影響を与えるため、ほとんどの場合、変異を起こした細胞は死滅するか、すくなくとも、集団の中で淘汰されてしまうため、遺伝子の進化速度は遅い。また、これらの遺伝子の変異が子孫に残るか否かについては、細菌のおかれた環境要因の影響はほとんど受けない。いわば、これらの ハウスキーピング遺伝子 の進化は、「進化の体内時計」として捉えることができる。MLSTでは、 ハウスキーピング遺伝子 を用いることが基本的考え方になっている。
※MLSTに関する詳しい解説ば下記記事でも解説しています。
※ハウスキーピング遺伝子 とその他の遺伝子の系統進化での適合性については下記記事に詳説しています。
遺伝子検査による微生物の同定
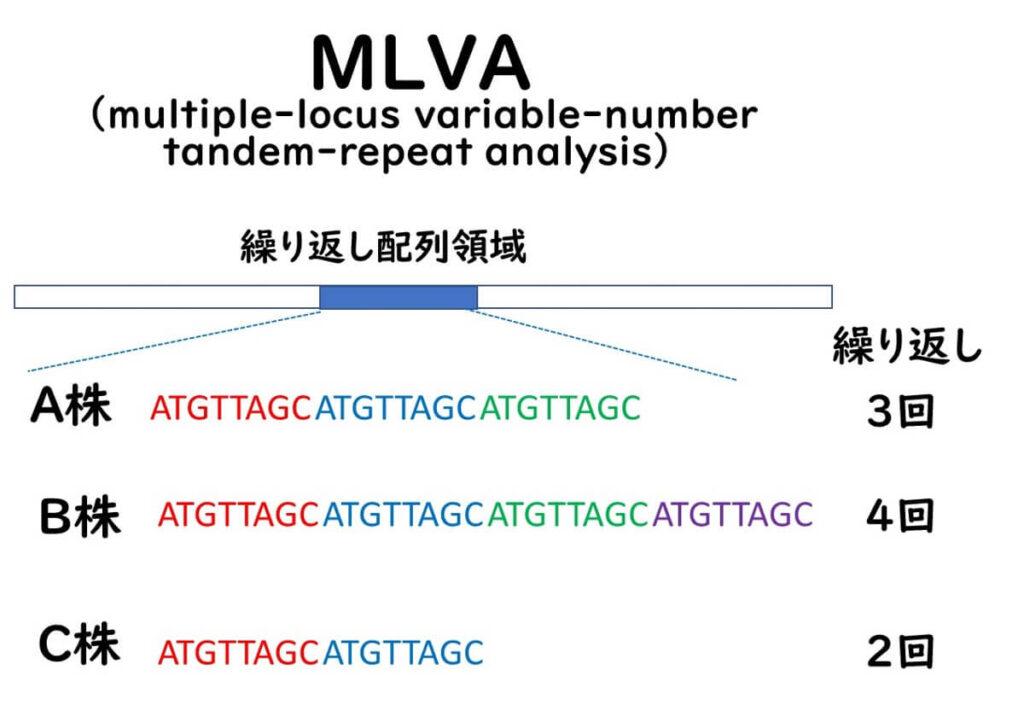
MLVA法(現時点での日本の腸管出血性大腸菌の分子疫学の標準法として採用)
MLSTの項でも説明しているように細菌の菌株の遺伝子型判別としてのパルスフィールドゲル電気泳動は、捜査の煩雑性やデータの客観性等において欠点を持つ。そこで、 MLS Tと同様に、細菌のゲノムが持つ繰り返し配列に注目して菌株の判別行おうとする方法として MLVA (Multiple-Locus Variable-number tandem-repeat Analysis) もある。登場時期はMLSTに少しおくれたが、だいたい2000年頃である。
MLVA (Multiple-Locus Variable-number tandem-repeat Analysis) 法は、細菌ゲノム全体に分散している反復DNA配列 (タンデムリピート)のコピー数を検出するものである。特定の遺伝子座におけるこれらのタンデムリピートの数は細菌の株ごとに異なる。このような遺伝子座ことの繰り返し数のこと形のことをVNTR( Variable-number tandem-repeat )とよぶ。VNTRは複数の遺伝子座または領域に存在する。
多くの研究が、細菌株の株レベルの識別法としてこれらのVTNRが適用可能であることが明らかになっている。特に、大腸菌O157などの非常にクローン性の生物の場合、MLVAは適した分子疫学的なツールであることがわかっている。
標的の細菌の種類ごとにあらかじめ定められた VNTRについてPCR プライマーによって、この繰り返し領域を挟むようにしてPCR増幅する。その後、PCR産物はアガロースゲルまたは自動キャピラリーDNAシーケンサーによって解析する。タンデムリピートの数は、PCR産物のサイズに基づいて判定できる。MLVAプロファイルはVNTR遺伝子座の繰り返し数によって定義される。MLVAプロファイルには、それぞれMLVAタイプ番号が割り当てられる。プロファイルコードは、株の比較や疫学研究のためにデータベースに保存可能である。
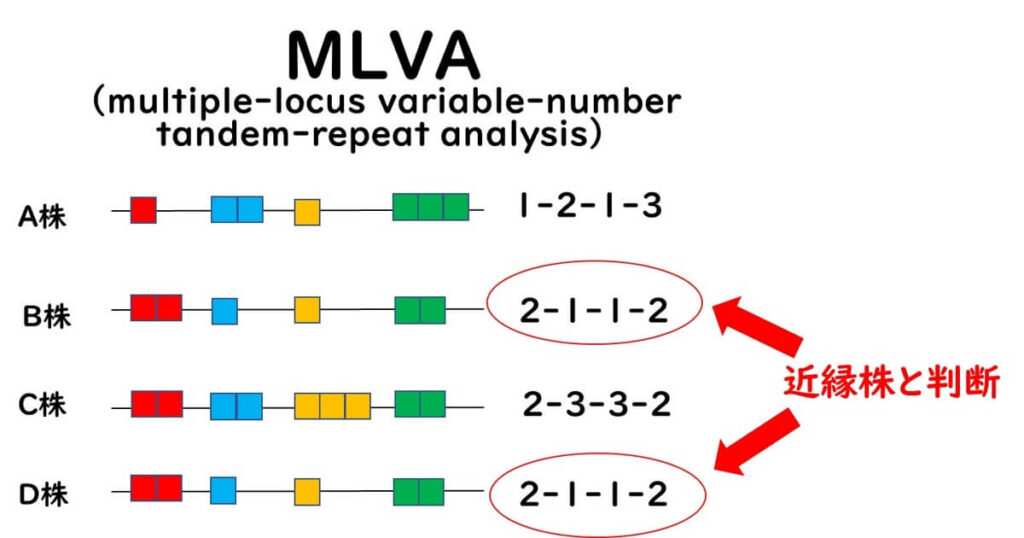
MLVA法は、特に、近縁の菌株間の遺伝的差異の検出が可能な解像度の高い手法である。したがって、特に、大腸菌O157などのクローン性の高い微生物菌株の場合、MLVAは適した分子疫学的なツールである。事実、日本では、腸管出血性の分子疫学的解析において、2018年からEHECの血清型O157、O26およびO111による食中毒の感染源特定の分子疫学的解析法として用いられている。 MLVA のデータベースを国立感染研究所と全国の地方衛生研究所で共有している。
このように MLVA はかなり近縁種に関しての分析学的解析ツールを、次世代シークエンサーを用いなくても、従来のサンガー法のキャピラリーシークエンサーで簡便にできる点で優れた方法ではある。しかし、すべての細菌で繰り返し配列があるわけではないので、 ML STほどには広く普及していない。また、公的衛生機関による食中毒細菌の分子疫学解析については、以下に述べるように、世界的には、次世代シークエンサーを用いた手法に移行しつつある。
全ゲノム配列(Whole Genome Sequence: WGS)にもとづく解析
2010年代に次世代シークエンサー(next generation sequencing: NGS)が登場した。次世代シークエンサーの登場により、従来のキャピラリーシーケンサー(サンガー法)に比べて、数千倍から数万倍の速度で遺伝子配列を決定できるようになった。これにより微生物菌株の全ゲノム配列(Whole Genome Sequence: WGS)にもとづく 食中毒細菌の分子疫学解析が現実のものとなった。全ゲノム配列解析の飛躍的解析能力の向上とコストの低下により、細菌の株タイピングの世界においても、もはやフラグメントではなく、配列解析を日常的に実施できるようになった。
生物、医学領域におけるDNA塩基配列決定のペースはあっと驚くようなものである。次世代シークエンサーの登場により微生物の分子疫学的解析の世界に更なる革命が起きている。
in silico MLST
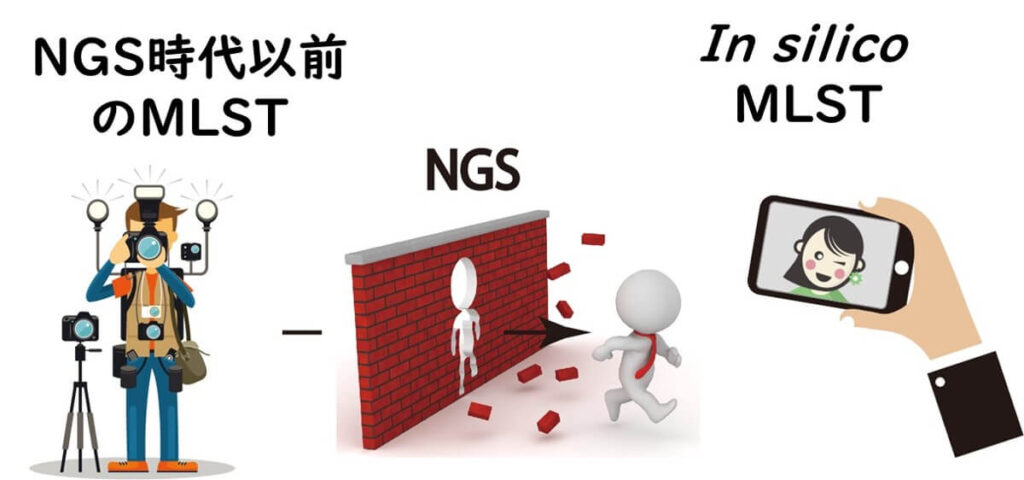
まず、上述したMLST法もNGS の登場により進化型が登場した。従来型のMLSTでは7遺伝子を個別にPCR増幅して塩基配列を決定していた。しかし、NGSの登場により全ゲノム配列をコンピュータ内で解析を行うin silico MLSTへと移行した。
注)in silico(イン・シリコ):「シリコン内で」と直訳される。意味としては「コンピュータ内で」を意味する。in vivo (生体内で)や in vitro (ガラス、すなわち試験管内で)などに準じて作られた用語。
2000年に登場したMLSTは上述したように、7つの遺伝子の配列をもとにしたタイピング方法である。過去20年間において、筆者の研究室も、この7つの遺伝子を一つ一つ抽出をして、それらの遺伝子配列を決めるという方法を用いてきた。しかし次世代シークエンサーが登場したことにより、この煩雑さは過去のものとなった。今では、分離した細菌を次世代シークエンサーで一括して分析を行い、その分析した DNA データをパソコンに入力して専用サイトにアクセスすれば、自動的に in silico MLST のタイプが出てくるようになった。手間暇は劇的に減っている。
また、in silico MLST 技術は、サルモネラの血清型の迅速判定法として英国公衆衛生研究庁(Public Health England)で採用されている。2015年4月に英国公衆衛生研究庁はサルモネラの血清型判別にWGSを導入した。従来の7遺伝子を用いたMLS T解析を血清型の同定に使おうというものだ。WGSのデータをコンピュータ内で解析しin silico MLSTを行う。この方法が導入されたのは、STによる血清型判別が実用的に使用可能と判断されたためだ。
※サルモネラ菌の血清型をin silicoMLSTで解析の詳細は下記別記事をご覧ください
サルモネラ菌の血清型を次世代シークエンサーで決定ー英国では2015年からin silico MLST法を導入しています
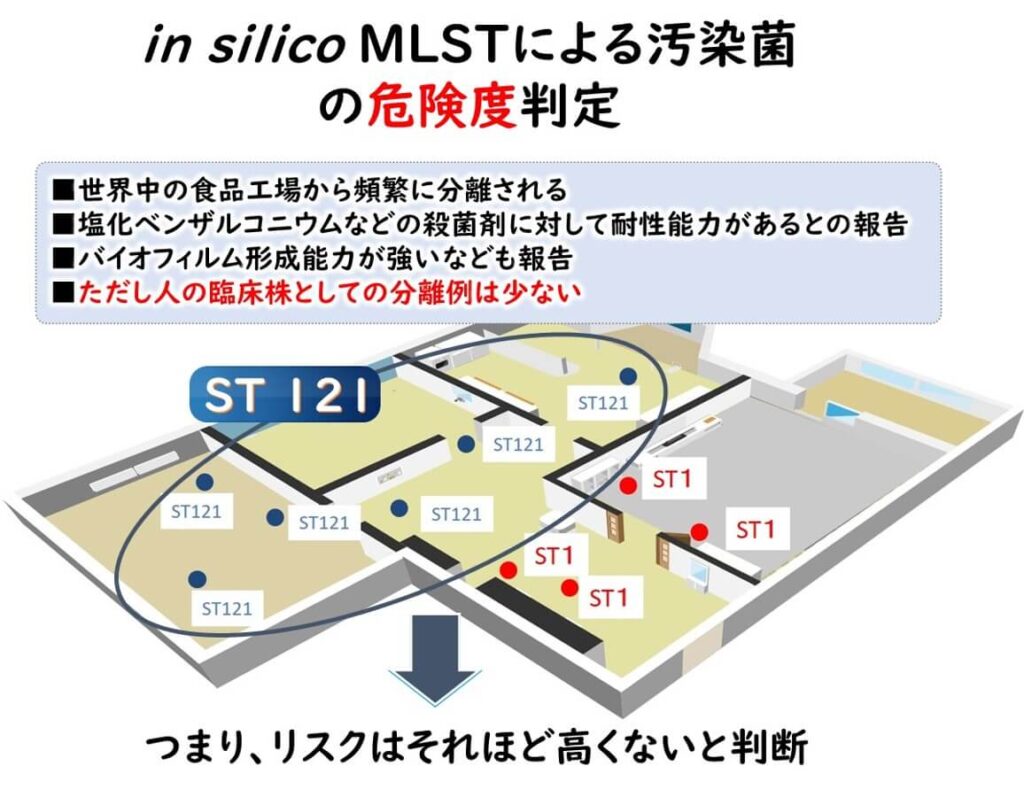
それでは具体的にこの in silico MLST で何ができるかを見てみよう。今仮に読者の工場でリステリア菌が頻繁に検出されているとする。分離株を in silico MLSTでタイピングを行なってみる。その結果、下図のように2つのタイピングの ST が出現したとする。
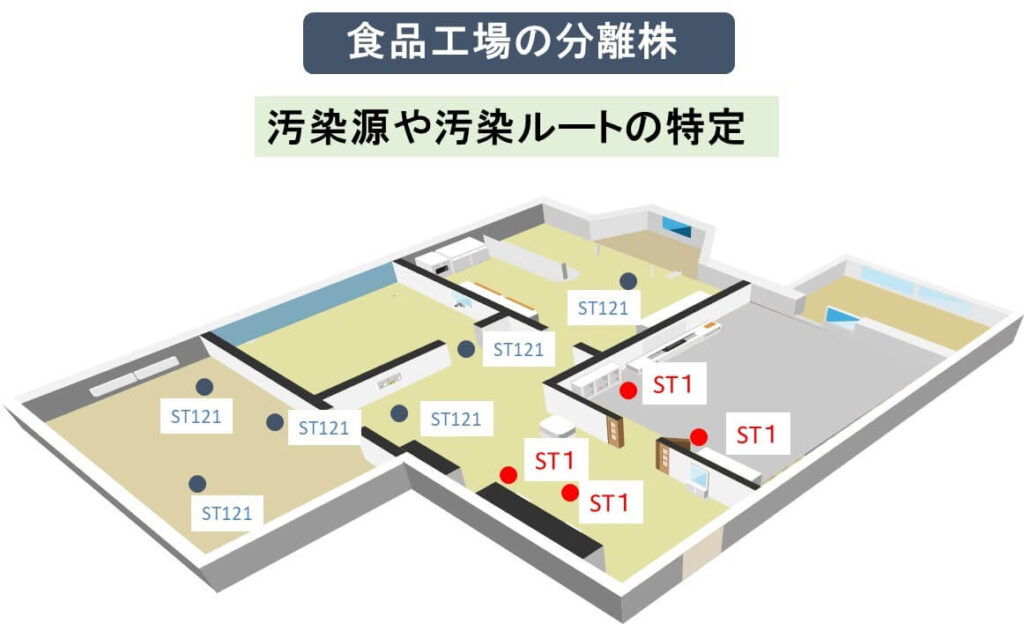
一つは ST 221だ。 ST 221は国際的なデータベースに照合してみると世界的に多くの食品工場で頻繁に検出されるが、それほど危険性の高くないリステリア菌だということがわかる。したがって深刻な心配を持つ必要はないということがわかる。
一方、 ST 1も分離されている。 ST 1はこれまで世界的にたくさんの死者を出した深刻な危険なリステリア株だということがわかりる。 読者の分離したリステリア菌が ST 1であった場合には直ちにこれを重大な心配と捉えて、工場を徹底的に洗浄と殺菌を行うべきである。
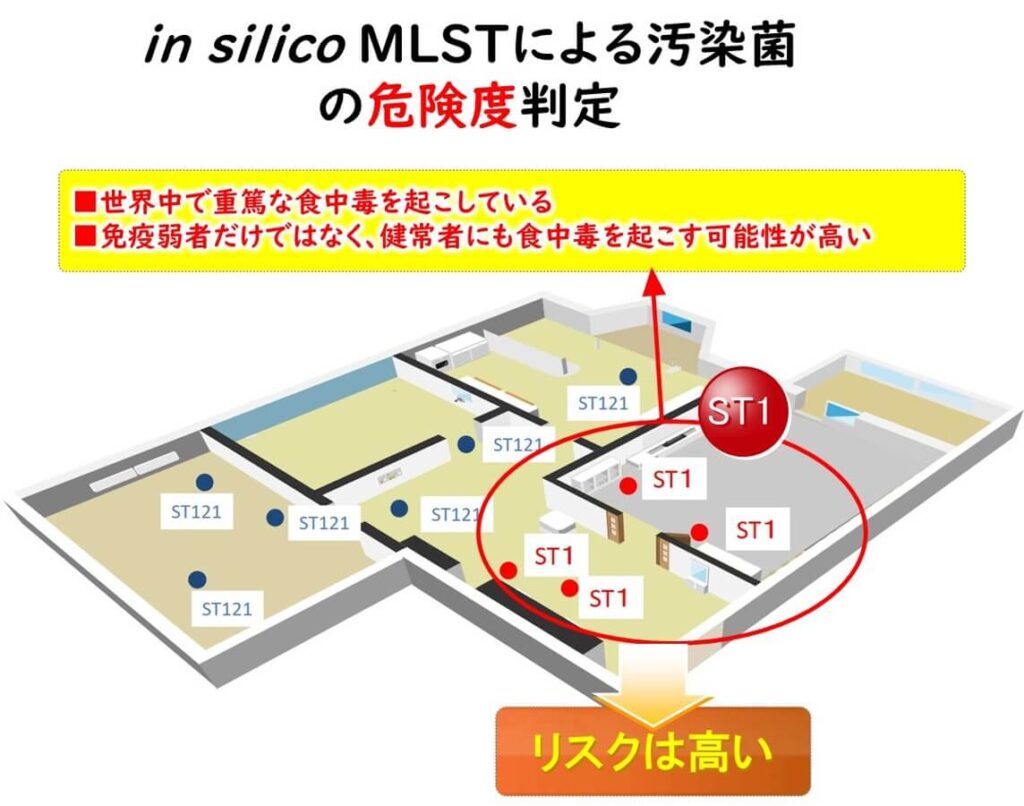
このように株のレベルで食品工場で情報を得ることによって有効な対策を打つことができる。これが in silico MLST の食品工場での活用法の一例だ。
※なお、in silico MLSTの活用法(菌種毎にどのようなSTがどのような特徴を持っているか)をもっと詳しく知りたい方は、下記総説に筆者がまとめているので興味ある方は御覧ください(英語)
Will the emergence of core genome MLST end the role of in silico MLST?
全ゲノム配列(WGS)にもとづく解析 を大別すると
さて、in silico MLST以外で、現在までにWGS解析で最も広く用いられている分子疫学技術は、次の2つに大別できる。
- 1塩基多型(Single Nucleotide Polymorphism:SNP )法
- Gene by gene 解析
NGS解析における細菌の分子疫学的解析手法

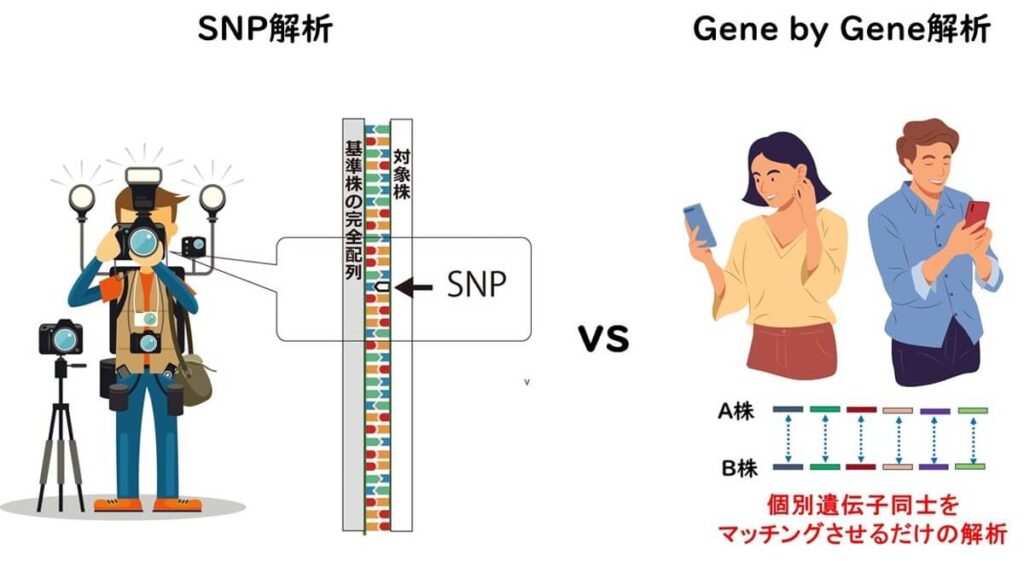
SNP解析
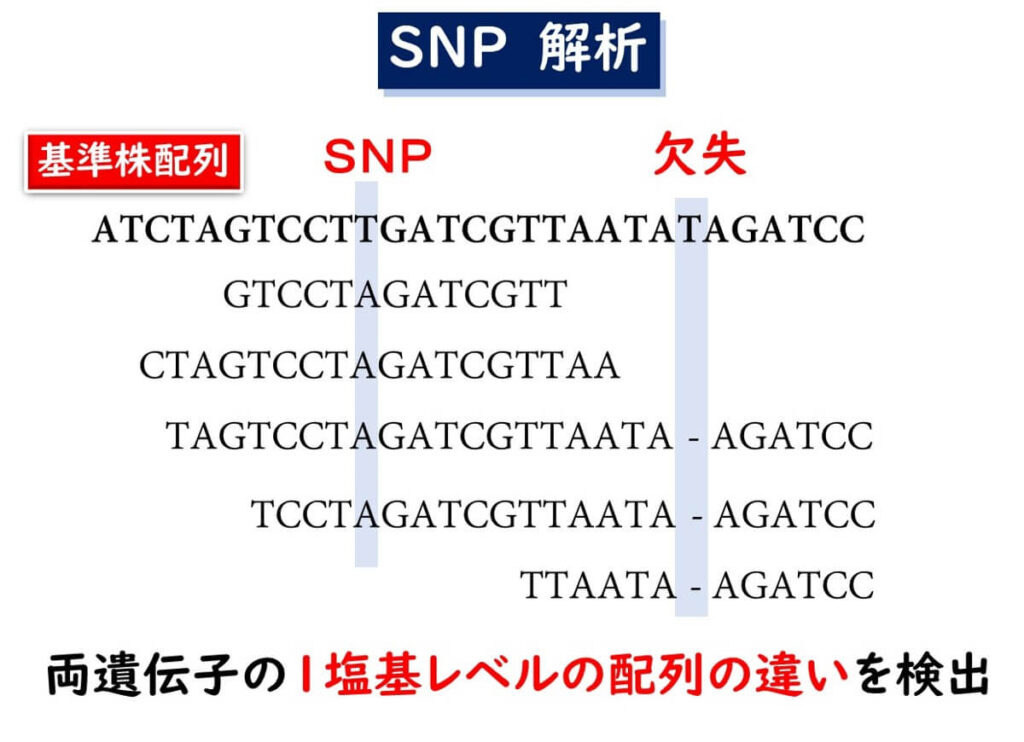
全ゲノムにおける1塩基レベルでの違いによって株の違いを識別する方法である。このような方法を1塩基多型(SNP,sinngle nucleotide polymorphism)解析と呼ぶ。snp解析では基準となる今株の遺伝子配列とのアライメントが必要となる。SNPは非常に情報量の多いマーカーであり、均質なグループの進化の歴史を明らかにできる。後述するように、過去数年間、多くの国でサルモネラ食中毒の分子疫学解析に用いられている。
基準株と1塩基でも塩基配列が異なる箇所があれば、その箇所をSNPとして認識する。また基準株に対して欠失している場合も同様である。
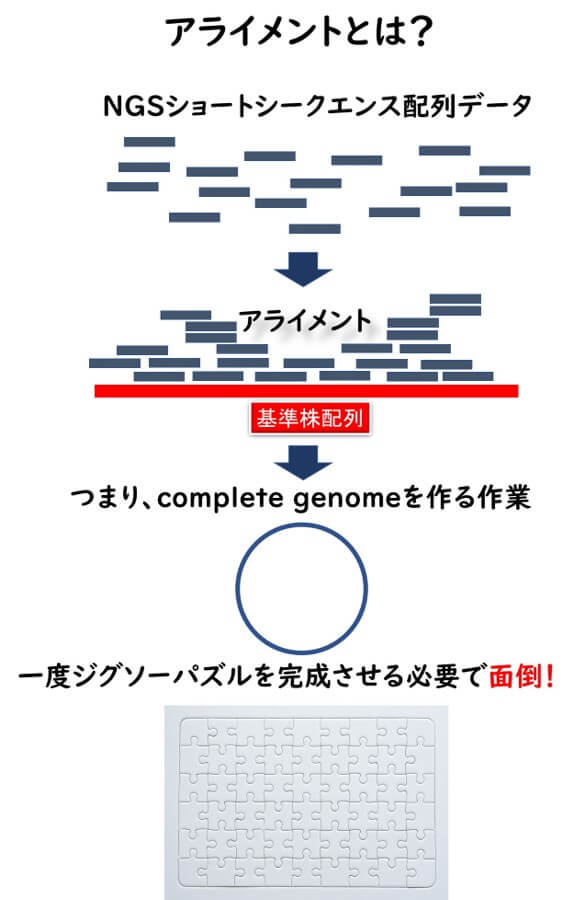
SNP解析においては、次世代シークエンサーで配列を決定したshort readのフラグメントを、すでに配列が決定されている基準株の配列に張り合わせていく作業が必要となる。このような作業のことアライメントと呼ぶ。SNP解析では、対象分離株の配列データを基準参照株の完全ゲノム(コンプリートゲノム)と正確にマッピング(1塩基ずつの照合)をしなくてはならない。次世代シークエンサーで配列の決定されたショートシークエンスの断片をジグソーパズルの一つ一つの断片に例えると、これらの断片を一度ジグソーパズルとして完成させないと SNP 解析が行えない。したがってとても作業が煩雑で時間もかかる。
そのため、SNP解析には高度なバイオインフォマティックスの知識と解析ツールが必要である。また、分析による使用基準株の違いは研究室間での標準化を困難にし、一貫性のある国際的なプロトコルの確立の障害となっている。
SNP解析の長所と短所
SNP解析は以下のような長所と短所を持つ。
【長所】1塩基レベルで解析ために解像度が極めて高い。
【短所】
1)基準株とのアライメントが必要(煩雑、長時間、性能の高いコンピュータ、bioinformaticsの知識が必要)
2)結果情報を他の研究者と共有しにくい(自分の対象とするアウトブレイクだけの解析には向いている)
3)相同組換えである遺伝子の水平伝播による影響で、菌株の距離を過大評価しやすい。

SNPの短所について
SNPは食中毒細菌の分子疫学解析を専門に行うプロで、且つ、bioinformaticsの深い知識を持っている解析者にのみ向いている。2022年現在においては、これらの解析には高性能なコンピューターも必要である。
したがって、今後多くの行政検査機関などがNGSを用いた食中毒菌の分子疫学解析行っていくためには、もう少し汎用性のある簡単なパイプラインや手法そのもの開発が求められている。

gene-by-gene 解析
2013年に、NGSによる細菌の分子疫学的解析ツールとして、画期的な論文が出版された。上述したMLS Tを提案したオックスフォード大学のメーデン教授よる提案だ。この手法は、次世代シークエンサーで得られたショートシークエンスの断片を基準株にアライメントをすることなく解析する方法である。すなわち、NGSで得られた微生物のゲノムを構成する遺伝子ごとについてコンピューター上で比較を行い、その上でいわゆるMLST解析行うというものだ。
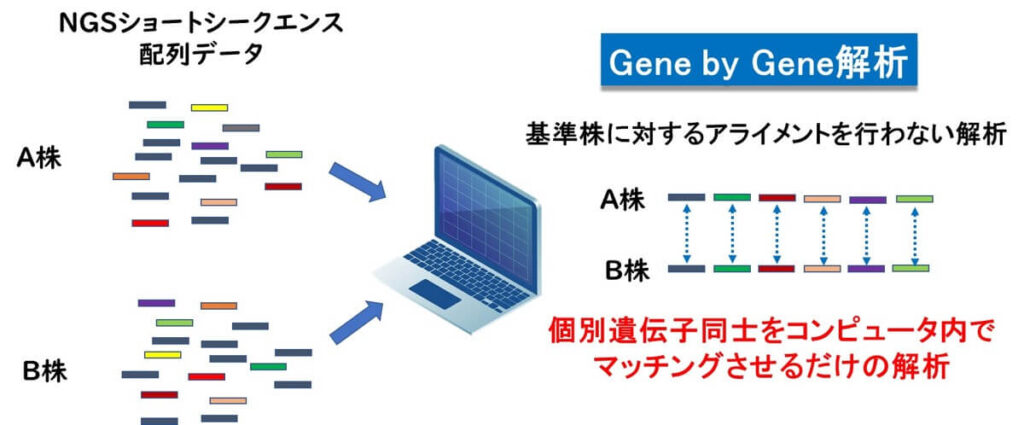
gene by gene解析では、
- 次世代―市区エンサーによって得られたショートリードのフラグメントをコンピュータ上である程度の長さに貼りあわせる。(このようにある程度の長さまではに合わさったDNAのことをcontigと呼んでいる)。基準株とのアライメントなど不要である(De novo解析)
- ひとつひとつの遺伝子ごとについて、分析の対象菌株同志の比較をコンピュータ上で行う。

gene by gene解析の長所
gene by gene解析には下記のような長所がある。
1)アライメントが不要である(高度なbioinformaticsの知識は不要)。
2)相同組換えや遺伝子水平伝播の影響を少なく解析することが可能である。
3)他の研究者とのデータ共有に優れている。
4)1つのアウトブレークだけではなく、別のアウトブレーク解析においてデータを解析し直すことも容易である。
以上に述べたようにgene by gene解析は ショートにリングの断片をバラバラにデータを出す次世代シークエンサーの特徴そのまま使って、簡便に全ゲノムデータのタイピングを行う方法と整理できる。その簡便さから、SNPよりも現場向きと考えられている。以下に、gene-by-gene解析の代表的な技術であるwhole genome MLSTとcore genome MLSTについて解説を加える。
whole genome MLST(wg MLST)
メーデン教授がgene by gene解析として具体的に提案した方法はwhole genome MLS Tである。この方法では、従来7遺伝子のみで行っていたMLS Tを、全ゲノムに存在している遺伝子すべてで行う。次世代シークエンサーの登場により、MLS Tをわずか7遺伝子に限定して行う必要はなくなった。そこで微生物の持つすべての遺伝子についてMLS Tに行うというのがメーデン教授の提案であった。
ひとつの微生物は約2000から3000の遺伝子を持っている。これらの遺伝子の配列情報全てを用いた MLSTである。 これまでの7つの遺伝子を用いたMLSTよりもはるかに解像度が高くなることは容易に理解できるだろう。

上記論文はPubMed Central(PMC)で無料公開されています。
ただしwgMLSTにも欠点がある。それは微生物の菌株の持つすべての遺伝子を対象とするために、菌株によっては、ある遺伝子を持っていたり、持っていなかったりするということがおきる。このような遺伝子をaccessory genomeと呼ぶ。accessory genomeも含んだ MLSTは解像度は極めて高いので、クローンに近い極めて近縁な株同士の識別時には大きな力を発揮する。しかし、ある程度菌株に遺伝的距離があり、これらの遺伝子を持っていない菌株を含んで解析する場合は、これらの菌株を同じ基準で評価することができなくなるという難点がある。
このようなwgMLSTの欠点を補うために登場したのが次に説明するcore genomeMLSTである。
Core Genome MLST(cg MLST)
細菌の様々な株が共通に持っている中核的な遺伝子、すなわちcome genome(おおむね 1500から2500程度の数の遺伝子)のみを対象とする技術をcore genome MLST(cg MLST)と呼ぶ。微生物の持っているすての遺伝子についてMLS Tを行うよりも、対象とする微生物のすてすべての株が共通に持っている遺伝子(=core genome)のみでMLS Tを行った方が、その結果の汎用性や拡張性が高い。このような考えに基づいて、cg(core genome)MLSTが2015年に登場した。
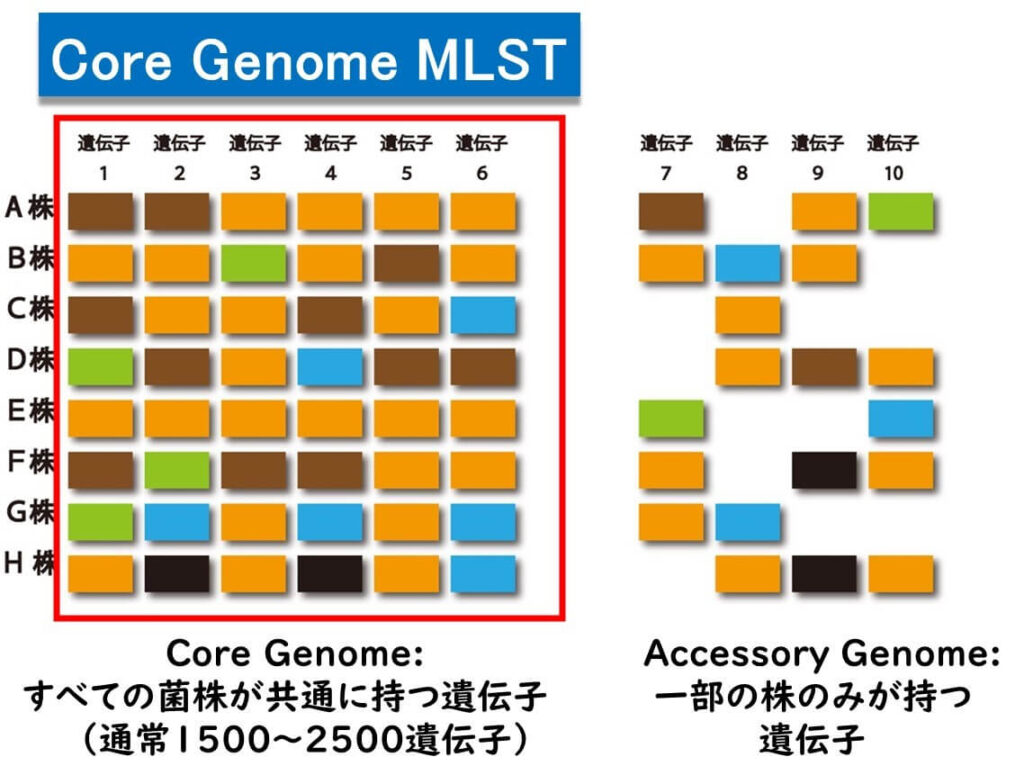
cg MLSTは、数百の遺伝子同志を独立してコンピュータ内で比較するだけの技術である。したがって、SNPのように基準株のコンプリートゲノムとのマッピング作業は不要である。NGSで得られる短い遺伝子断片配列をそのままコンピュータ内のみで解析できる。
2018年に、英国オックスフォード大学のMaiden博士(MLSTおよびcg MLSTの考案者) らのグループは、サルモネラ分離株の高分解能で再現性のあるタイピングを行うためのcg MLSTスキームを、ヨーロッパで発生したS. Enteritidisの大規模なアウトブレイクに適用して評価した。
その結果、cg MLST解析は複数の国でのS. Enteritidisのアウトブレイクを検出し、分離株のタイピングに十分な分解能を持っていることが示された。cg MLST解析の結果はSNP解析と一致していたが、cg MLSTでは保存されている共通遺伝子を解析に使用しているため、異なる研究室や管轄区域で容易に一貫して適用できるという利点が示された。
また、分離株数が多くても少なくても解析が可能であり、解析の再現性が高かった。
さらに、同じ研究室で、異なる時期に新たな分析を行っても過去の分析結果との前後互換性に優れていた。
加えて、ウェブベースの解析プラットフォームが利用可能になったことで、自前のパソコンへの高度な解析をバイオインフォマティクスツールのインストールを最小限に抑えて実施することも可能であることを示されている。
cgMLSTは今後20年間の分子疫学的ツールの主役になると予想される。
将来展望
次世代シーケンサー(NGS)技術の躍進は、微生物による食品安全の分野に新たな時代をもたらした。NGSの登場は、食品安全のための微生物検査において、約25年前に登場したPCRの登場のインパクトに匹敵するものだ。
食品由来の病原体のWGSを用いた分析技術は、MLST、MLVA、PFGEなどの既存の分子型別技術に代わって、すでに米国FDAやCDCなどで採用されている。この影響は、今後全世界に及ぶことが予想される。現時点では、SNPとcg/wgMLSTという2つの主要なアプローチが、食中毒原因食品の感染源解析の分子疫学的調査に使用されている。 どちらの手法も高い精度と分解能を有しており、今後も公衆衛生や規制機関の主要なツールとして使用されるであろう。

また、in silico MLSTやcgMLSTは操作が簡便なので、民間企業の食品工場での特定細菌の汚染ルール解析などでも用いられていくだろう。上述してきたSNPによるの分子疫学成果は、いわば、選りすぐられたプロ中のプロのバイオインフォマティクスの専門家たちの芸術的な仕事に例えることが出来る。これに対して、in silico MLSTやcg MLSTは高度なバイオインフォマティクスの知識がない実験者でも簡単に実施できる点が利点である。
食品業界におけるWGSの主な用途の一つは、汚染事象の根本原因を理解して迅速に対処することだ。そのため、WGSが日常的に広く使用されるためには、分析が便利で比較的迅速である必要がある。in silico MLSTやcg MLSTでは、オンラインデータベースを使用して、研究室間で容易に維持・共有できる点で優れている。これらの技術 は、2000年以降、食品民間企業でも活躍してきた16SrDNA と同じような汎用性拡張性を有している。cgMLSTについては、今後さらに多くの微生物でのデータベース化が進み、また、解析ソフトウェアの簡便化が加速すれば、今後20年間で、民間企業でも活発に用いられていくと予想できる。