本記事では食品微生物の遺伝子検査における PCR 法とはどのような技術か、リアルタイム PCR法とはどのような技術か、これらの技術の原理や 仕組み、利点を分かりやすく説明する。また、リアルタイム PCR の定量性について、臨床微生物分野と食品微生物検査においての応用の違いが出てくることについて説明する。
PCR法とその基本原理
PCR(Polymerase Chain Reaction)法とは、生物の特定の遺伝子をプラスチック製の小チューブ中で増幅する技術であり,検出したいDNA領域をはさむ特異的遺伝子断片(プライマー)を用意し,DNAポリメラーゼの鋳型特異的な合成反応を繰り返し利用する。ごく微量の鋳型となるDNAさえ存在すれば目的のDNA領域だけを短時間に10万倍から100万倍に増幅することができる。
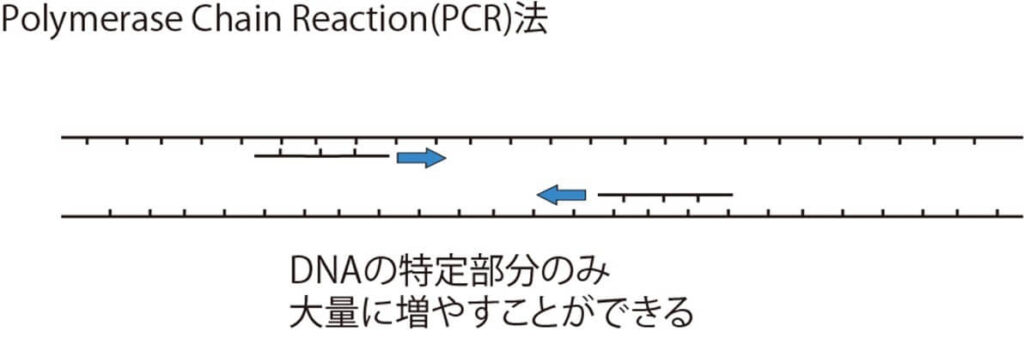
手順を簡単に述べると以下のようになる。
- まず、PCRチューブ(通常、0.6ml容量の小指の先ほどの大きさのプラスチックチューブ)に、未知サンプルDNA(食品から抽出した総DNA)、標的特定遺伝子のみに相補的な2本の増幅開始DNA断片(プライマー)、 遺伝子を増幅する酵素(Taq DNAポリメラーゼ)、および、DNA伸長の材料である混合ヌクレオチドを、それぞれマイクロピペットで入れる。
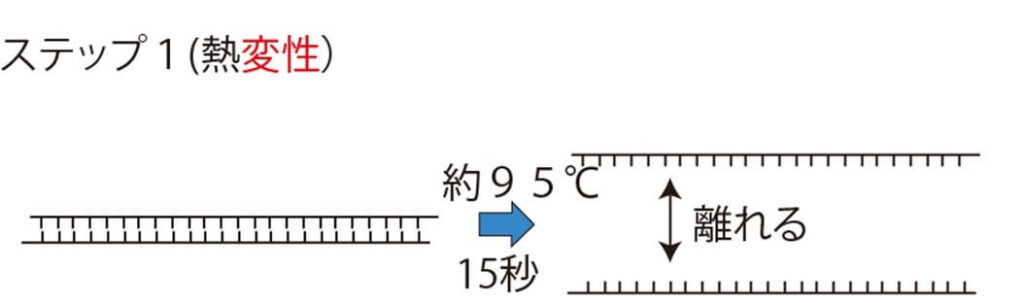
- DNAを熱変性させ、2本鎖DNAを1本鎖にする(90~95℃、約60秒 )。
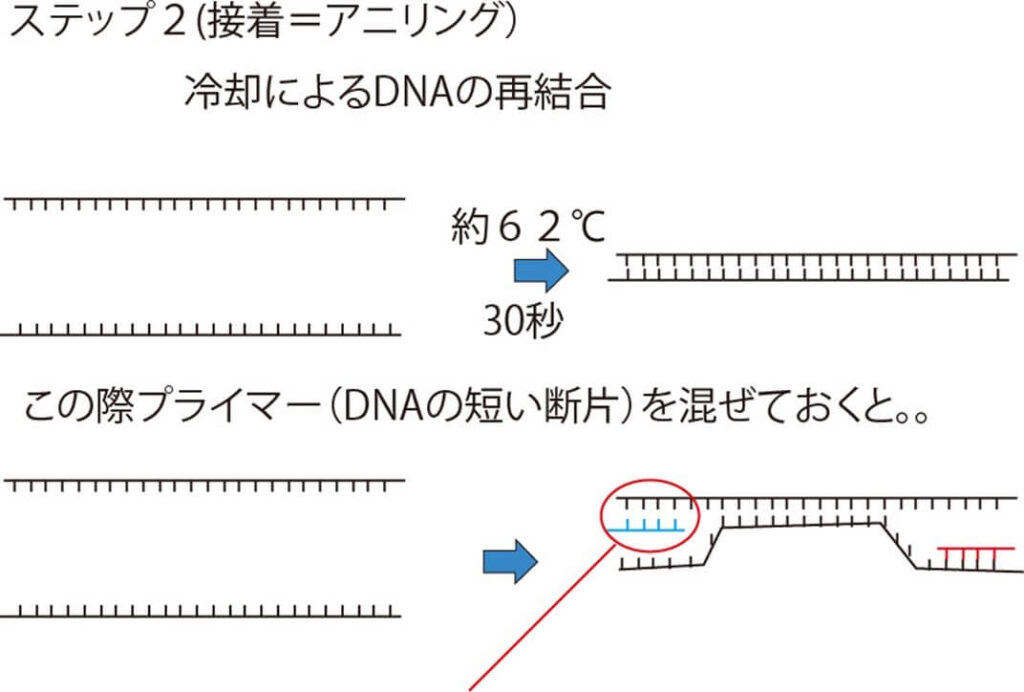
- 2本のプライマーを1本鎖になったDNAの標的領域を挟むように接着させる(アニーリング、40~60℃、約60秒) 。
なお、アニーリングという言葉の原語の英語の annealという動詞は、もともと、 冶金用語である。鋼・ガラスなどを焼き戻すという意味がある。PCR においては一度加熱した DNA を加熱して分離したものを冷却して再び二本鎖にすることが、この工程と似ているので、この言葉が使われている。
- Taq DNAポリメラーゼを用いて鋳型DNAの塩基配列に従って相補鎖の伸長反応を行う(70~75℃、約60秒)。
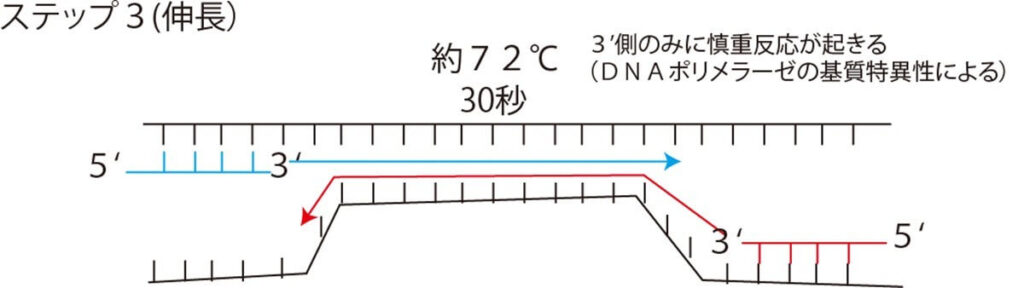
- 2~4の一連の反応サイクルに対応する3温度(条件により温度は多少異なる)をサーマルサイクラーの中で数10秒単位で繰り返し急速に変化させることにより、目的のDNA断片が指数関数的に増幅される。

細菌細胞の分裂では、その細菌の持っている遺伝子の全長(約106塩基程度)の複製後、新しい細胞の生育に必要なタンパク、脂質、炭水化物などの細胞構成成分の合成を行ってからはじめて細胞は分裂する。したがって、いかに増殖速度が速い細菌といえども、至適条件でせいぜい20~30分でやっと倍になるにすぎない。これに対して、PCR反応では、DNAを熱変性、アニリング、伸長の1サイクルに要する約3分間で目的遺伝子(通常数百程度の塩基数)が倍量になる。両者の差は指数関数的に増幅され、培養法では検出可能になるまで十分な細胞数を得るまでに最短でも1日かかるのに対し、PCR法では2~3時間で増幅遺伝子の検出が可能となる。
PCR法の原理を理解するためのDNAの性質
さて、そもそも DNAの基本的な性質を理解しておかないと、上に述べたような PCR の原理を理解できることはできない。以下に上に述べた PCR の基本的な性質を理解するために必要な DNA の特性について簡単に説明する。
なぜ、95℃と62℃のサイクルなの?
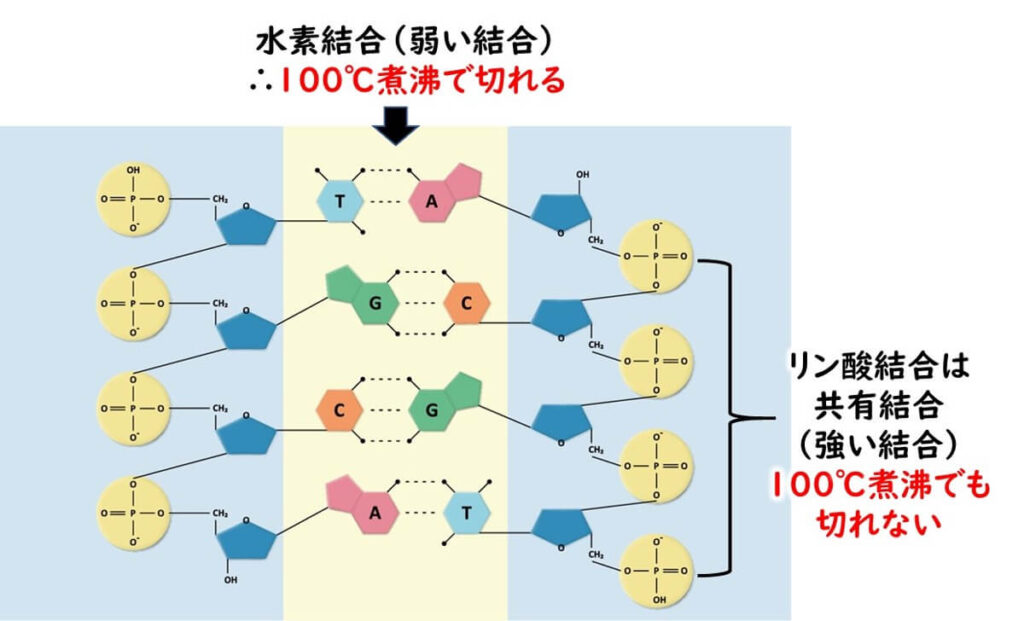
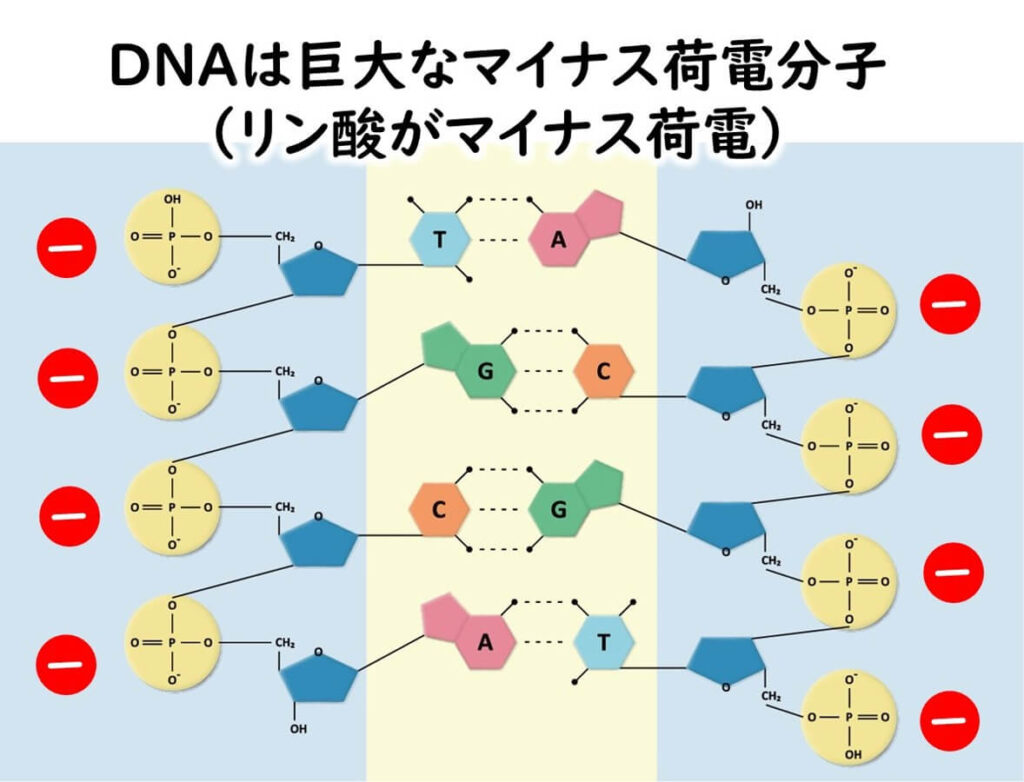
DNA は、デオキシリボースがリン酸結合を介して鎖上につながった構造である。そしてこの鎖が2本合わさって、はしご状の二重らせんを作っている。この鎖を合わせる、いわばジッパーのような役割を果たすのがアデニン、チミン、シトシン、グアニンの4つの塩基である。
この塩基同士の引き合いは、水素結合である。水素結合というのは、電子をお互いに共有するわけではない。互いの分子の若干の電荷の偏り(プラス寄り、もしくは、マイナス寄り)によって引き合う弱い結合である。このような弱い水素結合は100°Cのような煮沸条件では、水分子の急激な動きに耐えかねて、切れてしまう。したがって PCR 条件での95°という条件では DNA の2本鎖は一本鎖に剥がれてしまう。
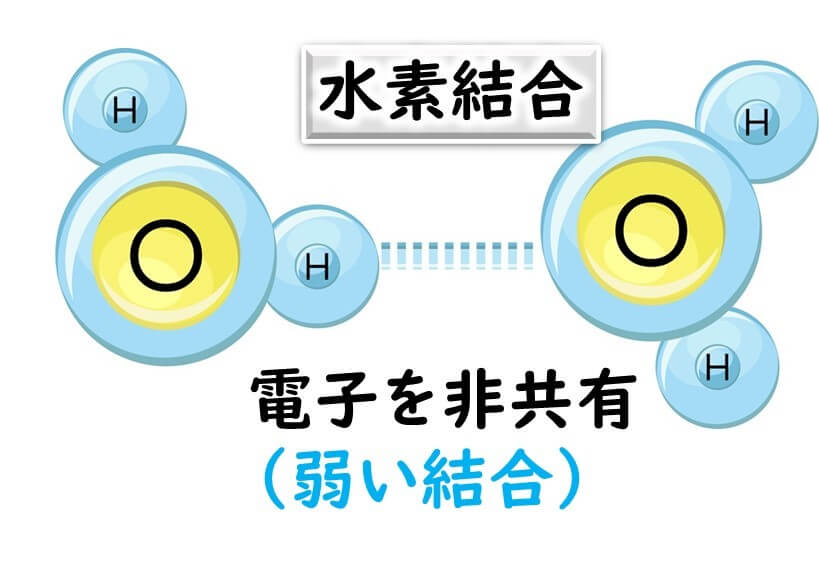
一方、デオキシリボース同士を結びつけているリン酸結合は共有結合である。共有結合とはお互いの分子の電子をガッチリとを交換し合って結合している。お互いの電子を共有しているので、そう簡単にはこの結合は切れない。

100°の煮沸条件ぐらいではこのような二重結合はビクともしない。従って PCR 条件での95°という変性温度ではこの鎖自体は切断されない。
以上のような DNA を構成している水素結合と共有結合がそれぞれ原因となって PCR 法の原理が成り立っている。
なぜ、3’側にしか伸長されないの?
上に述べた PCR 法の原理は DNA ポリメラーゼが DNA 断片の3’側だけを伸長する性質に基づいている。では、なぜ DNA ポリメラーゼは3’側だけを伸長するのだろうか?
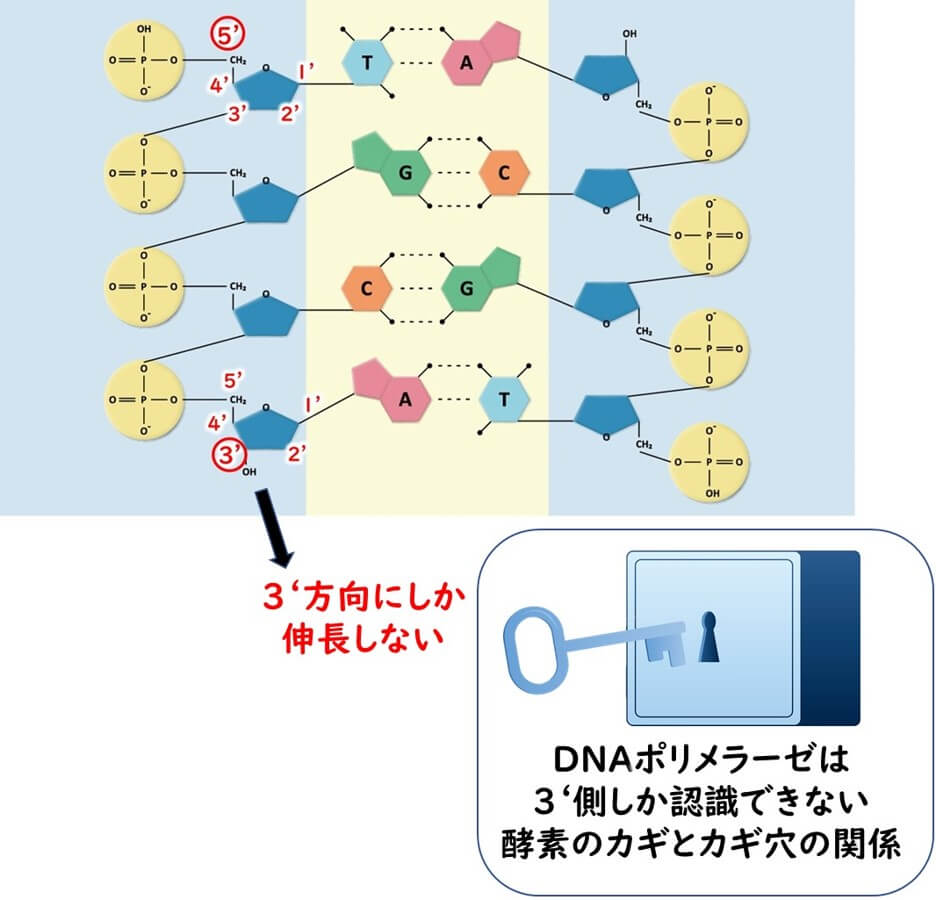
その理由は、酵素の基質特異性による。全ての酵素は基質となる標的物質と自身との構造との間に鍵と鍵穴の関係が存在する。 DNA ポリメラーゼもその例外ではない。 DNA を構成しているデオキシリボースを見てみよう。化学を学習した人はわかるが、分子を構成する炭素に番号を付ける習慣がある。 DNA を構成しているデオキシリボースについては1’から5’までの番号が右回りにふられる。なぜダッシュなのかといえば、 DNA の場合にはA,T,C,G の4つの核酸塩基を構成している炭素に1、2、3 などの番号が先にふられているからだ。これと区別するためにデオキシリボースの炭素番号にはダッシュがついている。
DNA ポリメラーゼは、酵素のカギと鍵穴の基質特異性の性質により、デオキシリボースの3‘側のみを認識する。5‘側の構造は DNA ポリメラーゼは認識できない。その結果 、DNA ポリメラーゼは3ダッシュ側のみを伸長するわけだ。
なぜ、72°Cという高温でDNAポリメラーゼ酵素が働くの?
PCR 反応においての DNA ポリメラーゼは95°Cの高温に置かれても失活しないことが求められる。しかし、タンパク質でできている酵素は65°Cを超えると失活する。
※タンパクの変性と熱との関係の基礎を確認したい人は下記の記事もご覧ください。
食品の加熱殺菌(パスツール殺菌)
現在の PCR 法が開発される前は、大腸菌の DNA ポリメラーゼを使っていた。大腸菌のDNAポリメラーゼはタンパク質の失活温度である65℃付近で失活してしまう。したがって、 大腸菌の DNA ポリメラーゼ では1サイクルごとに新たな新鮮な DNA ポリメラーゼを添加しないと反応が進まず、全く実用的ではなかった。
そこで登場したのがTaq polymeraseである。Taqポリメラーゼは米国イエローストーン国立公園の温泉中から分離されたThermus aquaticus という好熱細菌の持つ DNA ポリメラーゼである。この酵素の名前は、産生菌の属名の頭文字と種小名の頭二文字 (Thermus aquaticus) に由来する。この酵素は好熱菌の酵素なので PCR の条件下である65°Cと100°Cの間の温度帯でも失活しない。この酵素を用いる事によって初めて現在の PCR 反応ができるようになったわけだ。

PCR増幅の検出(電気泳動)
PCR反応により標的DNAが増幅されたか否かを判定するには、後述するリアルタイムPCR法が登場する20年前まではもっぱら電気泳動という手法を用いていました。もちろん今でも電気泳動は多くの実験室で用いられている。
電気泳動では、まず、PCR で増幅された遺伝子産物を寒天ゲルの穴の中に注入する。 DNA はリン酸結合で繋がった高分子化合物である。このリン酸基結合のリンの部分がマイナスに荷電している。したがって DNA 全体はマイナスに荷電している。
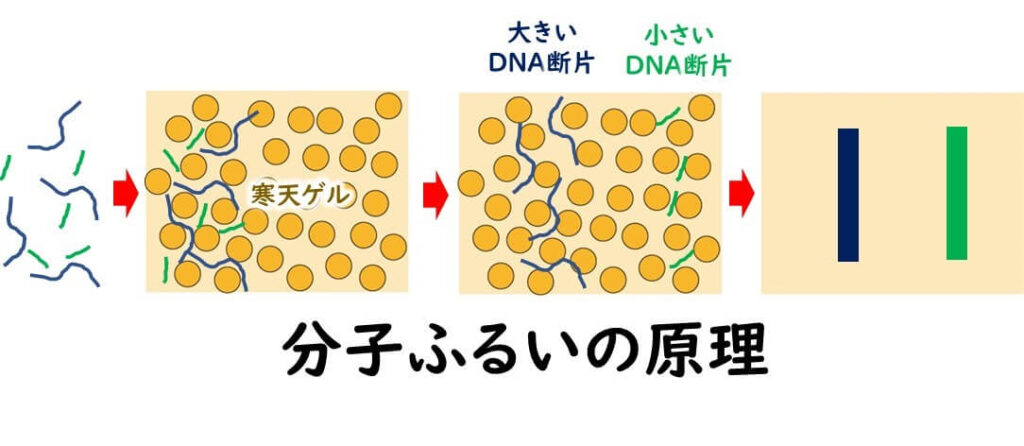
電気泳動ゲルのなかのDNAはプラス電極で引っ張られると、 DNA サンプルは寒天ゲルの中を移動していく。DNA の分子量が小さければ小さいほどゲルを簡単にすり抜けて素早く移動する。一定時間後、 DNA は寒天ゲル上にそれぞれの分子量に応じた距離を移動する。
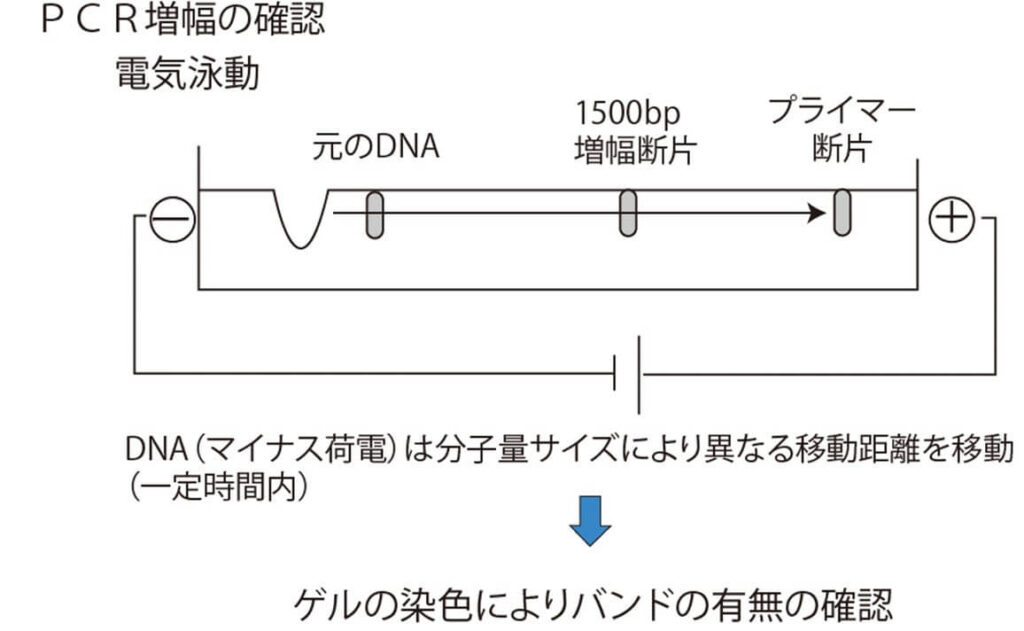
例えば PCR が目的とする遺伝子断片のサイズが1500bpだったとしよう。 1500bp に相当する距離に DNA が検出された場合には、 PCR で標的 DNA が増幅されたことを示す。 しかし 1500bp に相当するところに DNA が検出されない場合は、PCR 反応は陰性となる。これが電気泳動による PCR 反応の陽性と陰性の判定の原理である。
リアルタイムPCR法( TaqMan法 )
リアルタイム PCR 法とは PCR 反応が進むごとに反応チューブから蛍光を発する PCR のことである。反応が進むと蛍光量も増加する。この傾向値が一定の閾値を超えるまでの PCR サイクル数と元々の反応チューブにあった標的 DNA の量の間に 検量線を引くことができる。このようにしてサンプル中の DNA を定量することが可能となる 。一般的にはリアルタイム PCR と呼ぶことも多いが、正確にはリアルタイム定量 PCR が正しい用語である。以下では広く使われている用語としてリアルタイム PCR と呼ぶことにする。
様々な蛍光プローブが開発されている。その中でも最初に開発されたのがTaqManプローブ法である。この原理を理解すればその他の後発のリアルタイム PCR の蛍光プローブの原理もほぼ理解できる。したがってここでは TaqMan プローブ法について説明を加える。
リアルタイム定量PCR法(TaqMan法)がPCR法と異なる点は、PCR増幅の標的遺伝子を挟んだ2本のプライマーの間の増幅領域にさらに別の螢光プローブをアニーリング(接着)させる点である。
この際、プローブにはある仕掛けを仕組んでおく。すなわち、プローブの5’末端と3’末端に別々の色素を結合させておく。5’末端の蛍光色素(レポーター)は励起光を当てると蛍光を発する。一方3‘末端の色素は、消光色素(クエンチャー)とと呼ばれる色素である。5’末端側で発生した蛍光のエネルギーは、近くに消光色素が存在すると吸収されてしまう。結果として、いくら励起光をあててもプローブからは蛍光を発することができない。すなわち20bp~30bp程度の長さのプローブ上にレポーターと消光の2つの色素が存在している場合には、いくら励起光を与えてもプローブは蛍光を発することができない。
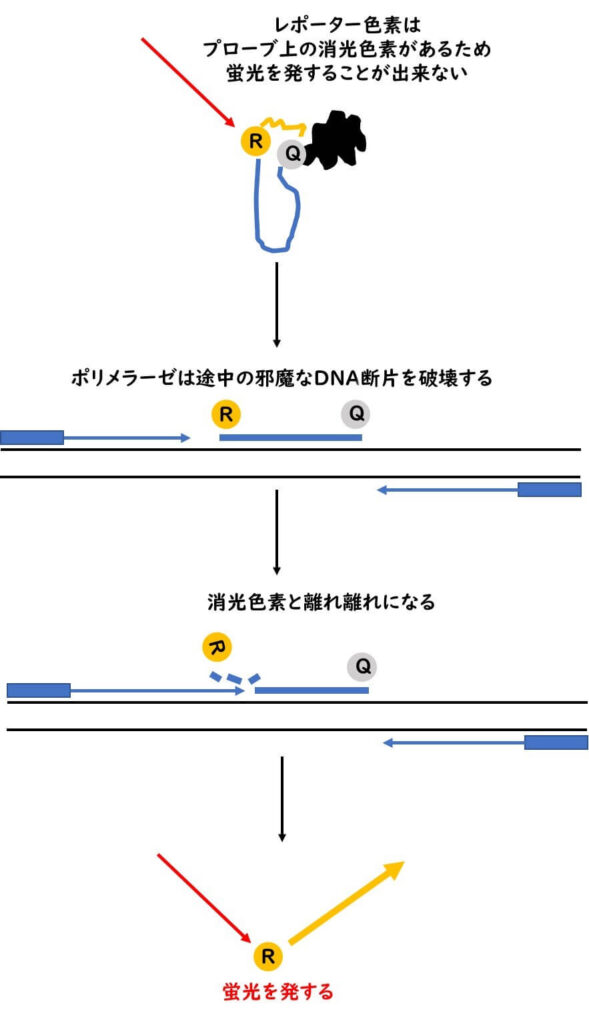
そこで、このプローブを PCR 反応の標的増幅領域の中間域に接着させる。 PCR 反応によって DNA ポリメラーゼが伸長反応を進めてきた場合、その増幅領域の途中にプローブが存在することになる。 DNA ポリ ポリメラーゼは進行中に邪魔者が存在している場合にはそれを1塩基ずつ切断するヌクレアーゼ活性を持っている。従って PCR 反応が進むことによって蛍光プローブは切断してバラバラとなる。その結果5'側のレポーター蛍光色素は3’側のクエンチャーから離れ離れになることができる。その結果、と蛍光を発することができるようになる。
2本のプライマー間増幅領域の途中にアニールした螢光プローブはPCR増幅とともにTaq DNA polymeraseの5'-3' exonuclease活性により切断され、結果として螢光プローブの発する螢光値が増大する。つまり、サンプル中に標的遺伝子が存在していれば、そのPCR増幅の有無が蛍光の発光として反映されるということである。
これを96穴プレート螢光光度計により測定し、PCR増幅反応の陽性・陰性を判定出来る。

リアルタイムPCR法の優れている点
リアルタイムPCR法がが優れている点は、次の3点に要約される。
(1)簡便性
いくら優れた技術であっても、技術の利用に熟練や高度な知識を必要とするのであれば、それは現場向きとは言えない。誰でもが、例えば、微生物教育を専門的に受けていなくても、1週間程度の研修で一通りの分析が出来、また、分析結果に個人差(習熟度)がないというのが理想的である。そういう点で、一般的なPCR法は、電気泳動ゲルの調製が素人向けとはいえず、また、発ガン物質であるエチジウムブロマイドを検出に用いるなど、各食品企業の現場検査担当者向けの技術とは言いがたい。そういう点で、 リアルタイム定量PCR法 は、サンプルをPCRにかけた後は、そのまま、蛍光光度計にチューブ毎装着し、分析ボタンを押すだけで結果が出てくるのて現場向きである。

(2)迅速性
電気泳動を省略しているので大量のサンプルを迅速に測定できる。したがって、現場向きである。PCR増幅後の反応液を96穴プレートに注入すれば96サンプルのPCR産物を15分で処理が可能である。96サンプルをこのように短時間で分析出来るメリットは食品現場では多大な力をもつ。96サンプルを手のひらサイズ(15cm四方)のプラスチック製ケース(96穴の少量サンプル挿入可能)一枚で分析可能である。
著者の実感として、大量のサンプルの検索をしたい場合などでは、一度TaqMan法を経験すると、従来の電気泳動依存型のPCR法が煩雑に感じられ、もはや従来型のPCR法には戻り難い感がある。

(3)定量性
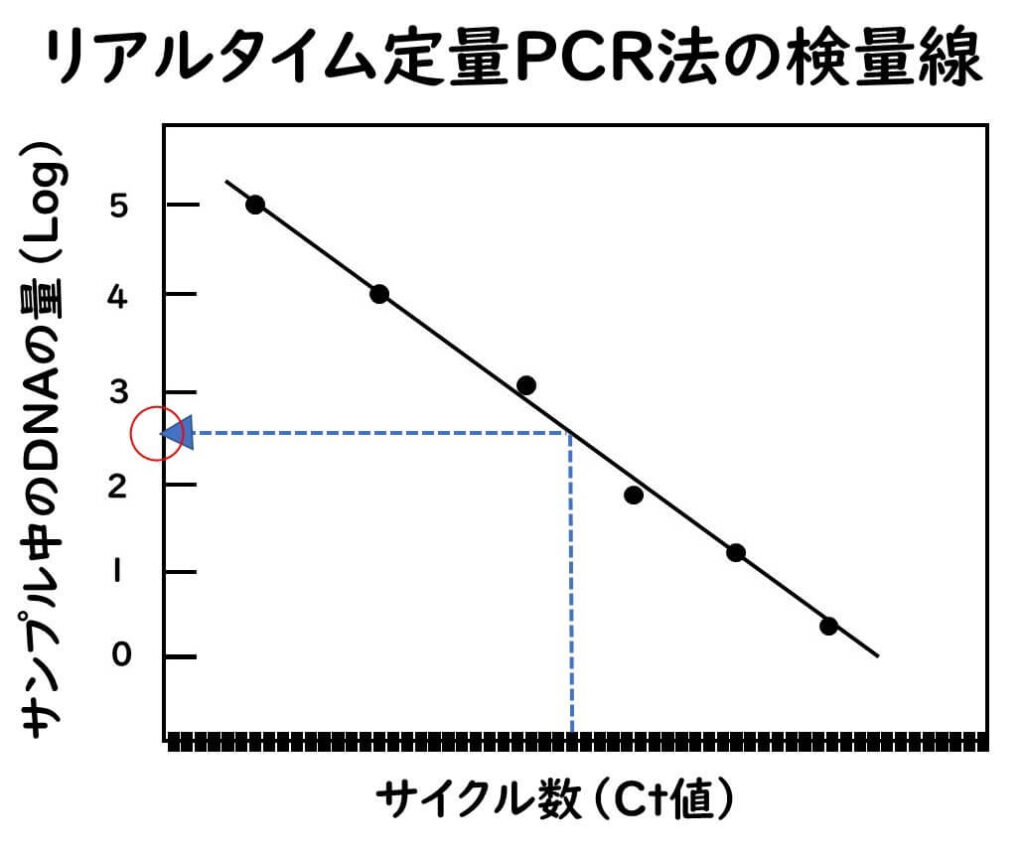
リアルタイム PCR が電気泳動に基づく PCR 法と異なる点は定量性があるということである。蛍光量が増加グラフで、ある一定のライン(閾値、threshhold)を超えるまでのサイクル数 (threshold cycle;Ct値) と、元々サンプルにあった 標的DNA量の間には逆相関が認められる。 Ct値) を横軸に、初発のDNA量を縦軸にプロットすることによって、検量線を作成することができる。
リアルタイムPCRと食品微生物検査
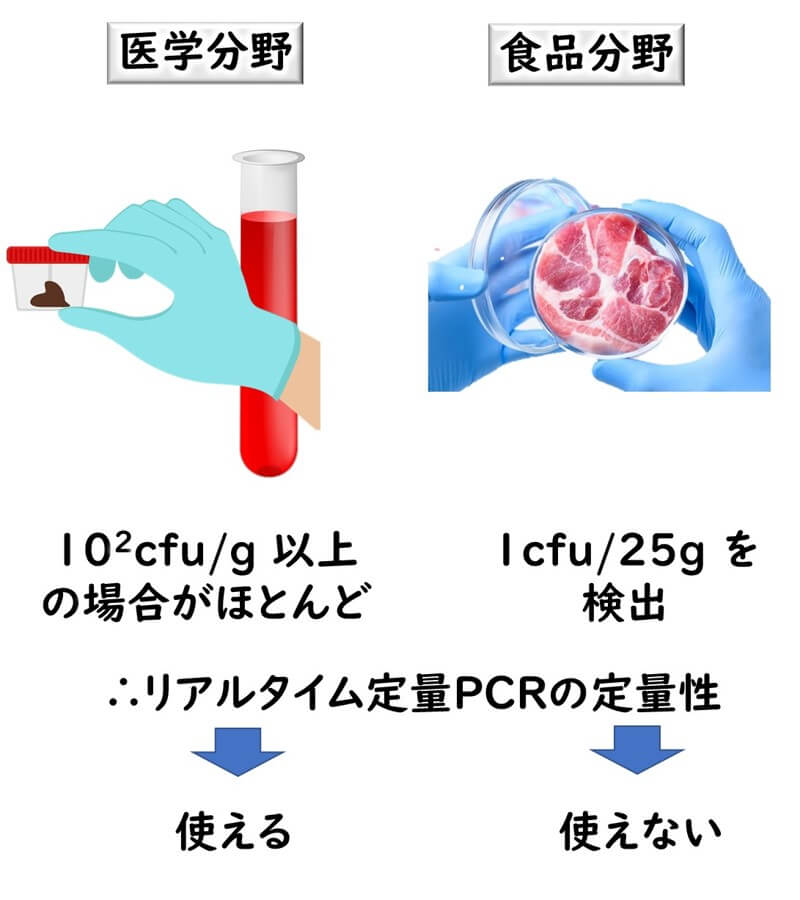
ただし、リアルタイム定PCR の定量性食品微生物分野で活用できているかといえば、答えは No である。その理由を以下に説明していく。
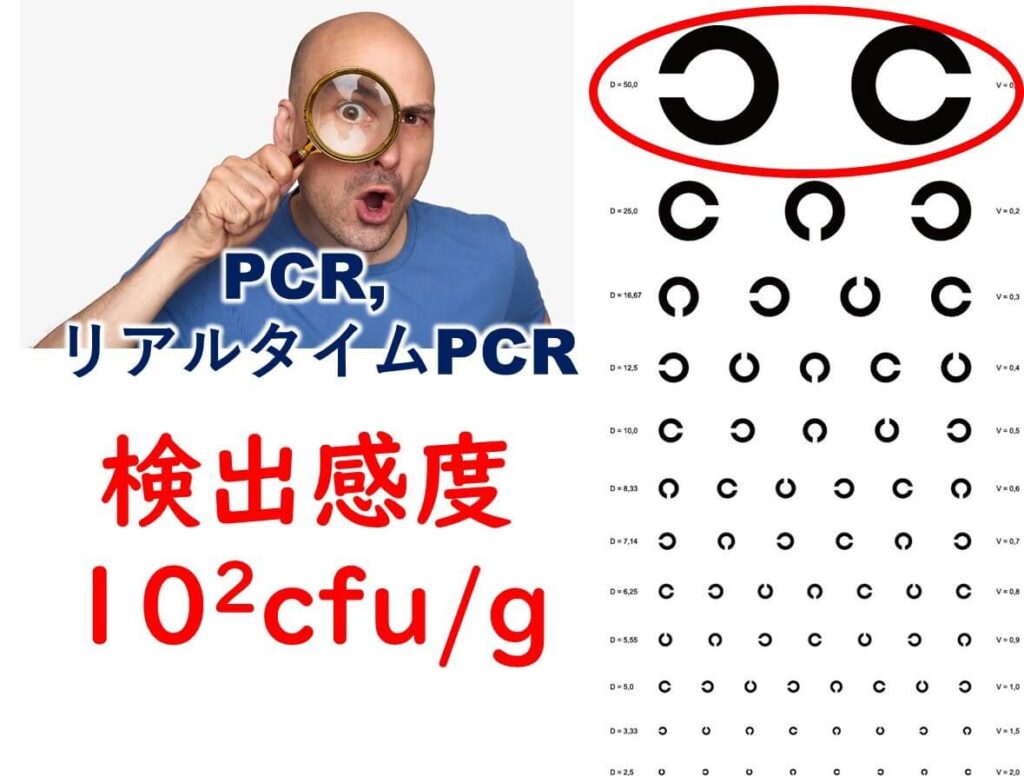
このことを説明するためには、リアルタイム PCR やそもそも PCR そのものをの検出感度の理解をしなくてはならない。最初に理解しなくてはならないのはリアルタイム定PCR や PCR の検出感度はサンプル中に1 g もしくは1ml に標的細菌が100cfu程度が存在しないと検出できないということである。なぜだろうか?
PCR やリアルタイム定量 PCR はプライマーの設計や反応系、および遺伝子抽出における食品からの阻害物質の除去がしっかりできていれば反応チューブの中に少なくとも1コピーの遺伝子が存在すれば増幅できる。その意味で検出感度はほぼ完璧である。問題は反応チューブ に標的 DNA を1コピー入れるまでの操作である。
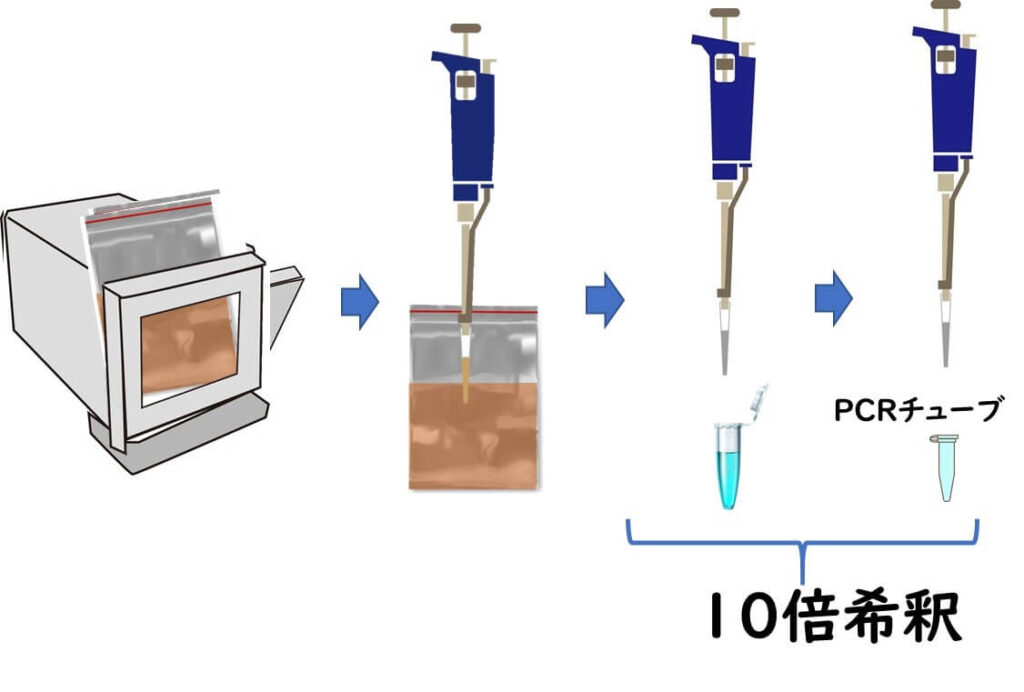
最終的な PCR 反応チューブに標的 DNA を1コピー出るためには、食品サンプル1 g中に100cfu存在しなくてはならない理由について、少し計算をしてみよう。例えば今1 g 中100cfuの微生物が存在していたとする。 DNA を抽出するためには通常お食品25 g を225mlの希釈水にいれ ストマッカー処理をする。 この段階で既に10倍希釈である。

またその後にサンプル駅から DNA を抽出する作業を行い、最終的に PCR 反応チューブにサンプルを入れるまでに、やはり10倍希釈程度の希釈がかかる。い遺伝子抽出法には様々な抽出法があるが、いずれにしても、この程度のサンプルからの希釈は最終的には避けられない。従って PCR チューブに少なくとも1コピーの DNA を入れるためには、逆算すればやはりサンプル中の1 G には100cfu以上が存在していなくてはならないことがわかる。
しかるに食品業界において 求められる検出感度は25 g に1cfuである。世界の多くの食品の微生物基準は25g あたりで存在するかしてないかが問題される場合がほとんどである。つまり 1g当たりで換算すると0.04cfuの検出感度が求められることになる。つまり、食品から直接 DNA 抽出して PCR をかけても食品業界で求められている検出感度には到底及ばない。

そこで食品の微生物検査にはどうしても増菌培養が必要となる。この点が医学/臨床微生物分野と根本的に異なる点である。

医学や微生物の臨床分野では患者の糞便中や血液中の感染菌の量を測定する。患者の感染状況によっては通常ほとんどの場合糞便中や血液中に1ml あたりもしくは1 g あたりに100cfu以上の標的感染菌が存在している場合が多い。あるいはそのような状況(感染状況)を検出することを目的としている。 一方、食品微生物の微生物検査では、医学臨床分野と異なり、食品中にわずかな食中毒菌が存在するかしないかの予防的な措置で検査を行う。したがって、このような高濃度の標的菌を検出するケースはほとんど存在しない。上述したように25 gに1cfuの標的食中毒菌が存在しているか否かを検出する。
微生物はいったん増菌培養液で増殖を開始すれば、もともと存在していた微生物量を正しく反映して増殖するとは限らない。つまり一度サンプルを増菌培養を行った時点で定量性は失われる。
以上、食品の微生物検査においては、リアルタイム PCR の定量性についてはほとんど活用する機会がない。リアルタイム PCR で反応とともに生じる蛍光が一定量を超えるか否かのポジネガ判定のみで実用的には運用している場合がほとんどである。つまり、リアルタイムPCR の蛍光のリアルタイム測定でポジティブネガティブを判定できるという利便性を活用しているということになる。

以上のように、 リアルタイム PCR の定量性については、このように医学領域と食品微生物領域では、その有効性が異なることを理解しておく必要がある。