PCR検査による食品微生物の検査は、迅速性という点で培養法に比べ優位である。ただし、 PCR検査 の技術特性と実施に伴うリスクについて正確に理解しておかないと、思わぬ落とし穴に落ちることになる。本記事では、PCR検査による食品から微生物の直接検出における留意点について解説する。
食品の微生物検査において遺伝子検査を用いる目的
本記事を読むにあたり、まず、PCR法とはそもそもどのような技術かについては、下記の別記事に詳細に説明したので、ご覧いただきたい。

PCR法を用いれば、食品から直接微生物群の遺伝子をまとめて抽出して、その中に目的細菌がいるか否かを検査できる。すなわち培養法を省略できる。
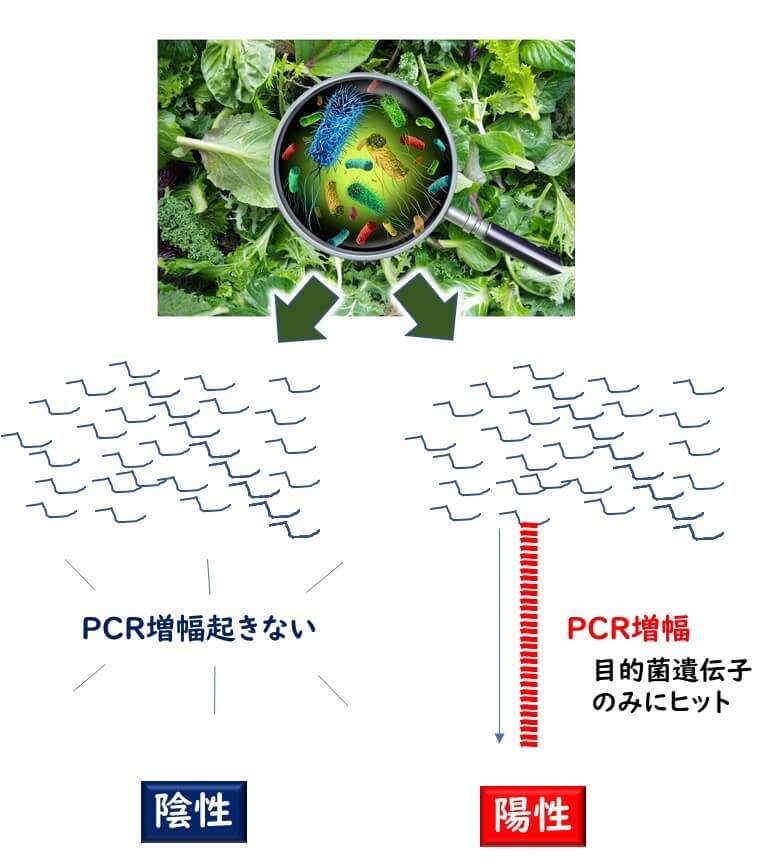
PCR検査法の最も大きなアドバンテージは、培養法に比べてはるかに速いスピードで結果が判定できるという点にある。この迅速性という利点は、大量のサンプルを処理して生きるという利点にもつながる。

従って、PCR法は別記事でも説明したように、培養法で標的菌の分離を行う前に、大量のサンプルを処理をすることによって、可能性を絞り込むというツールとして有効だ。
PCR法の食品微生検査における位置づけ(入門編)については、下記の記事にわかりやすく整理しているので御覧いただきたい。
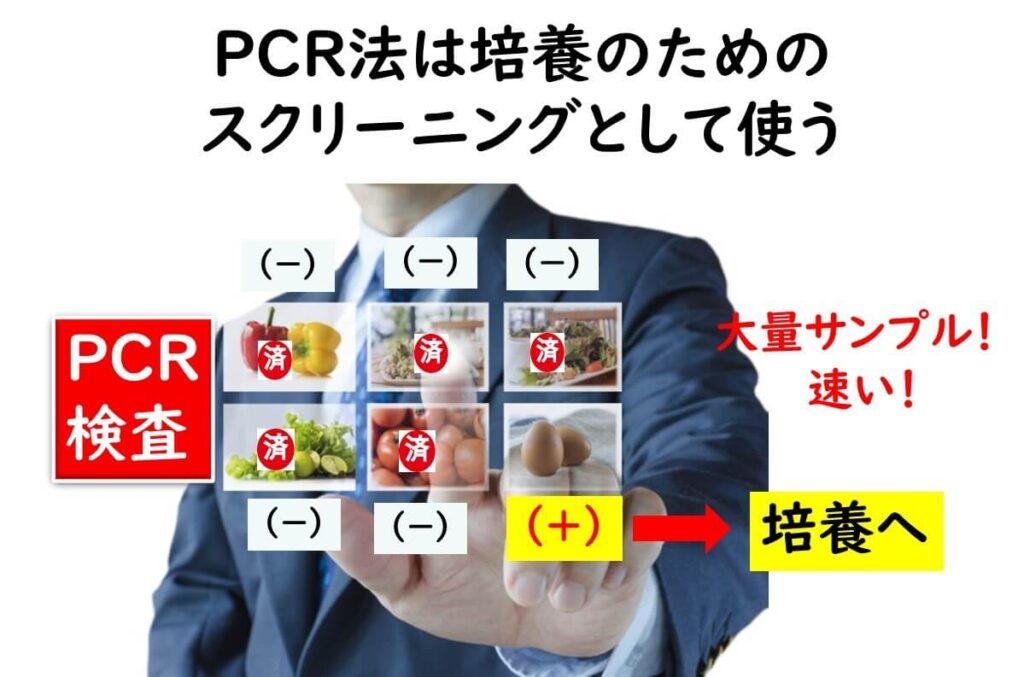
PCR検査のリスクと留意事項
上述したように、PCR法は、対象とする食品中に対象とする微生物が存在するか否かを確認するための方法として優れている。

上手に使いこなせれば、っ食品微生物検査において、強力な味方となる。
ただし、どのような技術もそうであるが、使い方を間違えると思わぬ落とし穴に落ちることになる。
PCRのの技術特性と技術に伴うリスクについて正確に理解して、使う必要がある。
以下に、これらについての主なものについてピックアップして述べる。技術特性の理解、サンプリングと増菌培養、遺伝子の抽出から増幅の各段階に分けて、主なポイントを述べることとする。
技術特性の理解
PCRのプライマーの性能
PCR反応において目的とする特定の遺伝子配列のみを増幅させるために、伸長の起点となる遺伝子断片を標的遺伝子に結合させておく必要がある。この遺伝子断片のことをプライマーと呼ぶ。PCR反応においては、二本鎖のDNAに、増幅したい遺伝子配列領域を挟むようにして、それぞれにこの起点となる遺伝子断片を結合させる必要がある。そのためプライマーは上流と下流にそれぞれ逆方向へ向けて2種類が必要となる。
PCR検査による食品から微生物の直接検出は、対象の微生物だけが持つ遺伝子を検出することが理想的である。これまでにほとんどの食中毒菌で、毒素遺伝子や病原性に関与する遺伝子などを標的としたプライマーが開発され、検出方法が確立している。しかし、分子マーカー(その菌だけが持つ特定の遺伝子配列)が見つけにくい菌群については、検出法の確立していないものも多い。対象菌のみが持っている遺伝子でなくても、類似菌との遺伝子配列の違いにより、対象菌を識別する場合もある。
例えば、下の図では、筆者らの研究室でかつて魚介類のヒスタミン生成菌(グラム陰性菌)のヒスタミン生成酵素(ヒスチジン脱炭酸酵素)の遺伝子を明らかにし,おそれらのPCR検出法を開発した事例であるが、下の図でアミノ酸配列がヒスタミン生成菌の間で保存されている領域(黒塗りつぶし)をプライマー領域とすれば、これらの菌のPCR検出が可能である。
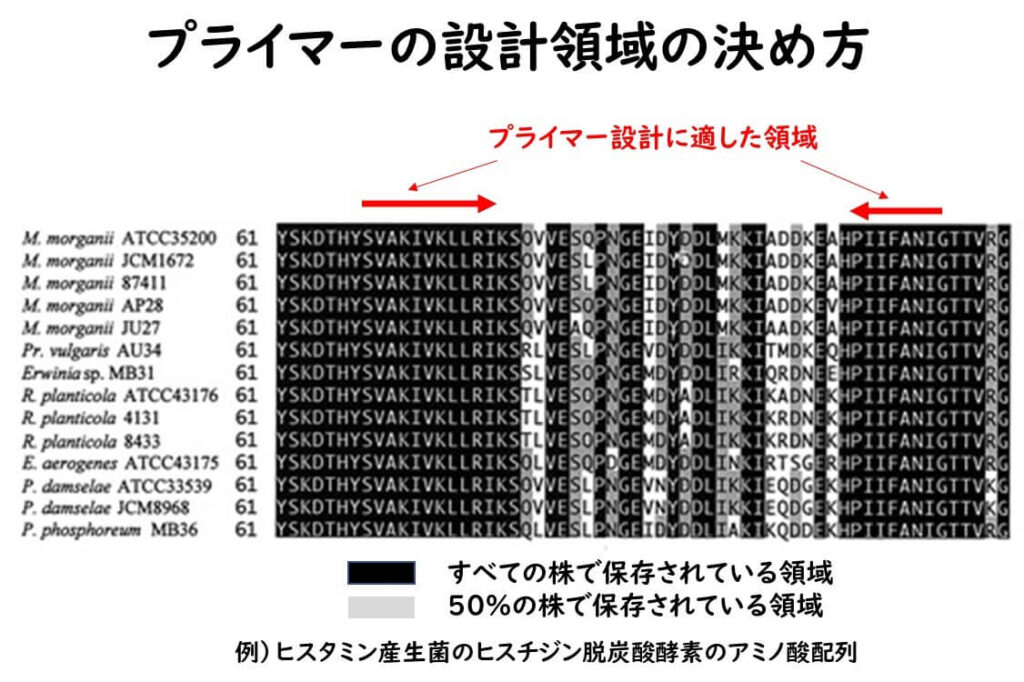
このように、食品の微生物検査において、自分でオリジナル対象菌のPCR検出法を開発するためには)、まずは、対象微生物の分子マーカー遺伝子の配列を決定することが不可欠である。自分で決定しなくても、データーベースから分子マーカーとなり得る遺伝子配列をダウンロードして、上記のように、比較する作業を行う。
プライマーの長さ
PCR反応系を自分で作る場合は、プライマーの長さについての基本事項の理解が必要となる。
プライマーの長さは、18bp~24bpに設計しなくてはならない。
なぜだろうか?

まず、プライマーの長さが短い場合について考えてみよう。
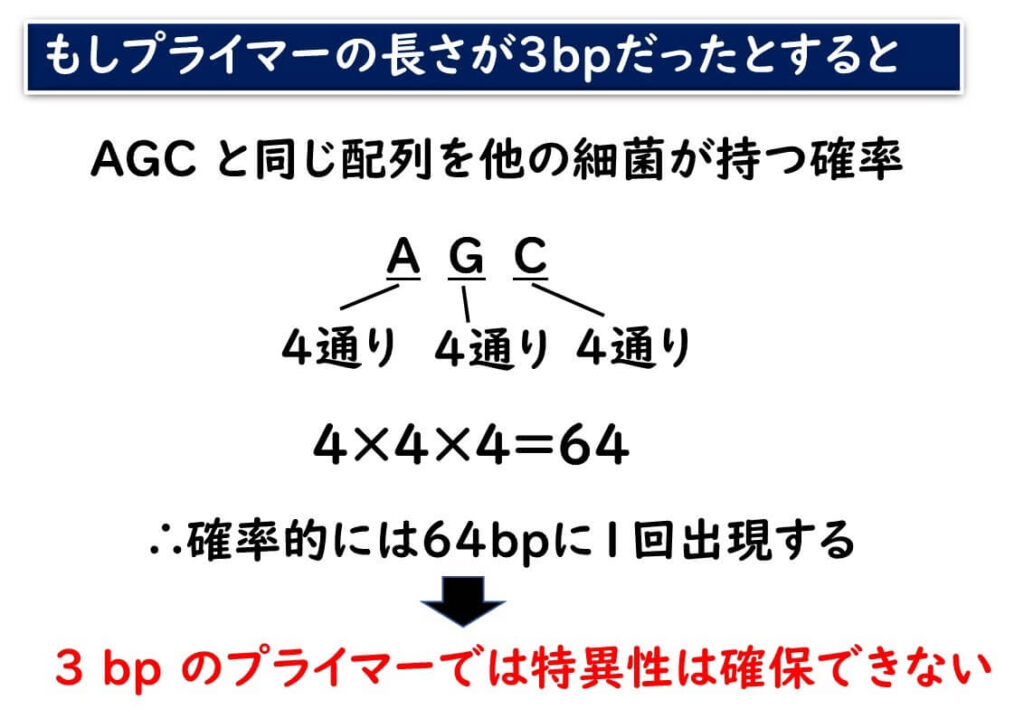
下の図のように、仮にプライマーの長さが3bpだったとしよう。単純に確率計算を行うと、AGCと同じ配列が出現する確率は、
4×4×4=64 となる。
つまり、64塩基に1回はこの配列が、確率的に生じることになる。これでは、ほとんどすべての細菌がこの配列を持つことになる。
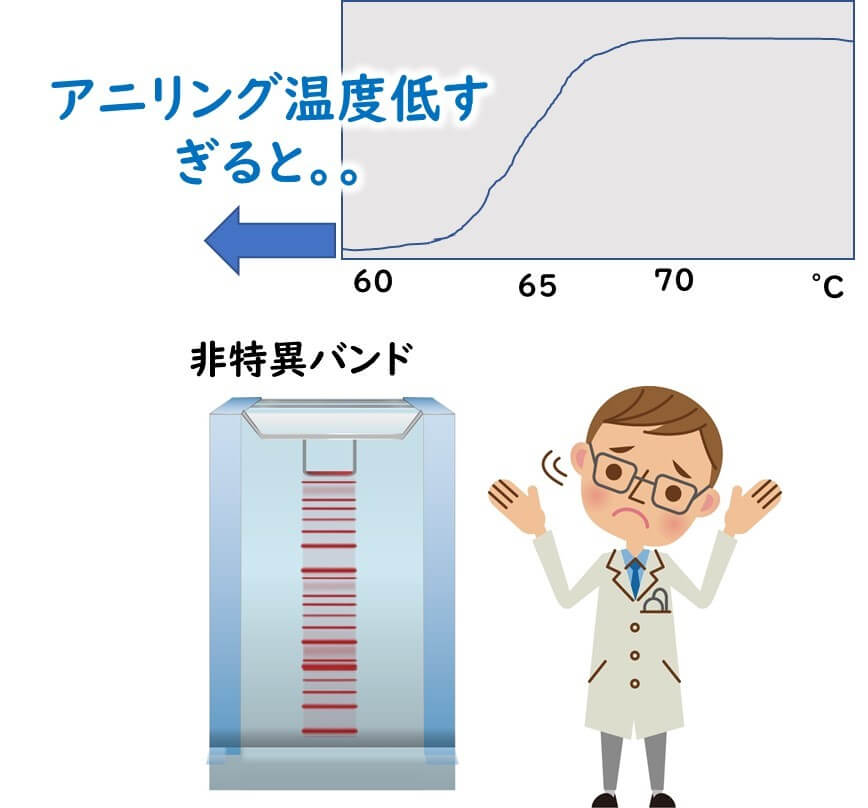
仮にこのような短いプライマーでPCR反応を行ったとすれば、下図のように、非特異バンドだらけとなる。

一方、プライマーの塩基数を20bpに設定した場合はどうだろうか?
この配列が偶然生じる確率は
4×4×4×4× 4×4×4×4× 4×4×4×4× 4×4×4×4× 4×4×4×4× 4×4×4×4=420=1012
となる。
しかるに、一般的に細菌の全ゲノムサイズは、 106 bpである。つまり、上記の配列が他の細菌で偶然生じる確率は 106 の1であるとわかる。つまり、 プライマーの塩基数を20b に設定すれば、検出の特異性が確保できる。

さらにフォワードとリバーズのプライマーで、それぞれに上記と同様の特異性を確保するので、検出の絞り込み精度はさらに上がる。

ただし、上記の計算はあくまでも、単純に確率論的に整理しただけである。実際の微生物の遺伝子の場合、上記のヒスタミン生成遺伝子でも述べたように、類似遺伝子が存在し、また、それらの遺伝子配列が標的菌の分子マーカーに近い場合も多い。したがって、プライマーの塩基数を20bpにしたからといって、完璧に特異性が確保できない場合も多い。
では、さらに、特異性を確保するために、プライマーの長さをさらに長くすればどうなるだろうか?

プライマーを長くしすぎると次の2つの欠点がある
- アニーリングに時間がかかり、PCR反応が遅くなる
- プライマーが2次構造を形成する可能性が高くなる
したがって、24bp以上の長さのプライマーはPCR反応では不適である。

Tm値とアニーリング温度
PCR反応においては、熱変性過程で二本鎖に解離した標的DNAがアニリング過程で再び結合する。この際、標的領域の配列を伸長するための起点となるプライマーも標的DNAに結合する必要がある。
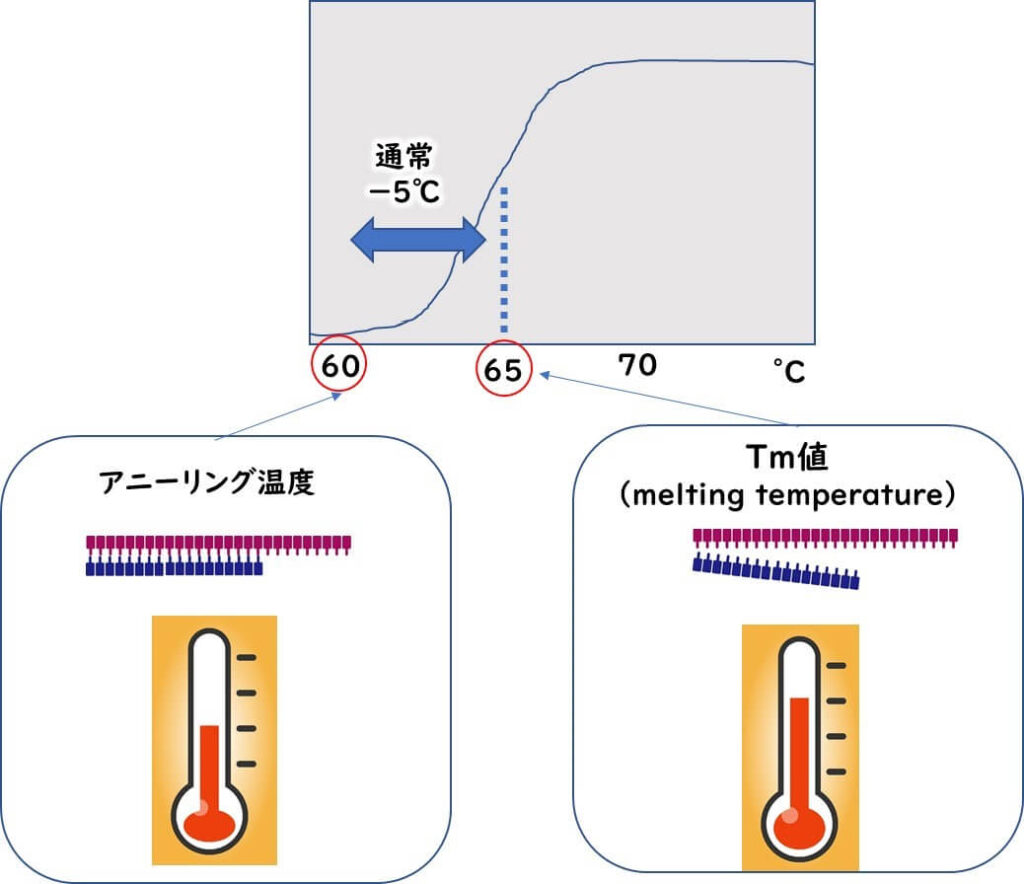
- PCR反応においてプライマーと鋳型DNAが接着するために設定される温度をアニーリング温度という。
- また、プライマーが標的配列と50%の割合で2本鎖を形成する温度ことをTm値(Melting Temperature 、融解温度)と呼ぶ。PCR反応に於けるプライマーを設計においてはTm 値は重要な要素となる。
標的配列に相補的なプライマーがアニーリング温度できちんと接着させるには、温度を低くすれば低くするほどしやすくなる。しかし、アニーリング温度を低くしすぎると、プライマーが非特異的に鋳型DNAへ接着していまう。結果として、非特異バンドが大量に出現することになる。
したがって、アニリング温度は低すぎてもダメである。
一般的にアニーリング温度はプライマーのTm値より5℃低くすればよい。
アニーリング温度でフォワードとリバースの両プライマーがきちんと標的DNAに過不足なく接着するためには、両プライマーのTm値が同じでなくてはならない。

したがって、プライマーの設計時には、単に遺伝子領域の保存性のみならず、その配列のTm値を計算した、両プライマーのTm値に著しい差異がないようにしなくてはならない。
では、Tm値はどのように計算するのか?
いろいろな計算式があるが、ここでは、もっとも簡便な手計算法のみを紹介する。
Tm値の計算法:
GもしくはC:4℃を与える
AもしくはT:2℃を与える
たとえば、下図のような配列ならば、
2℃+4 ℃ +2 ℃ +4 ℃ +4 ℃ +2 ℃ +4 ℃ +2 ℃ +4 ℃ +2 ℃ +4 ℃ +2 ℃ +4 ℃ +4 ℃ +2 ℃ +2 ℃ +4 ℃ +2 ℃ +4 ℃ +4 ℃ =62℃
Tm値は62℃となる。

GC 含量
プライマーと標的DNAの結合の安定性については、プライマーのDNA配列のGC含量も重要となる。
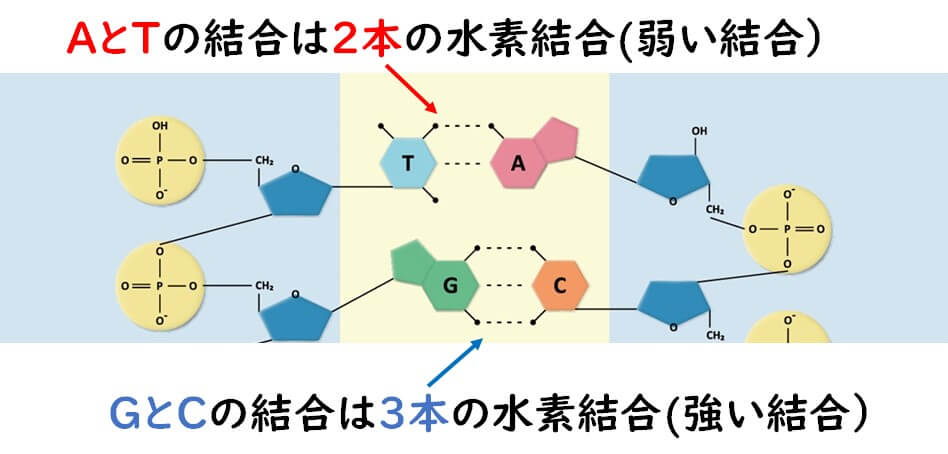
生物のDNAの配列はアデニン (A) とチミン (T)、グアニン (G) とシトシン (C)の4つの塩基により構成される。 二本鎖のDNAでは互いにAT対が二つの水素結合を形成する。これに対し、GC対は三つの水素結合を形成する。そのため、GC含有量が大きい配列では二本鎖のDNAは熱変性等により解離しにくくなる。
上述のTm値の簡易計算で、なぜ、GやCに4℃を与え、AやTには2℃を与えるかといえば、これはこれらの塩基の結合力の強さによる。
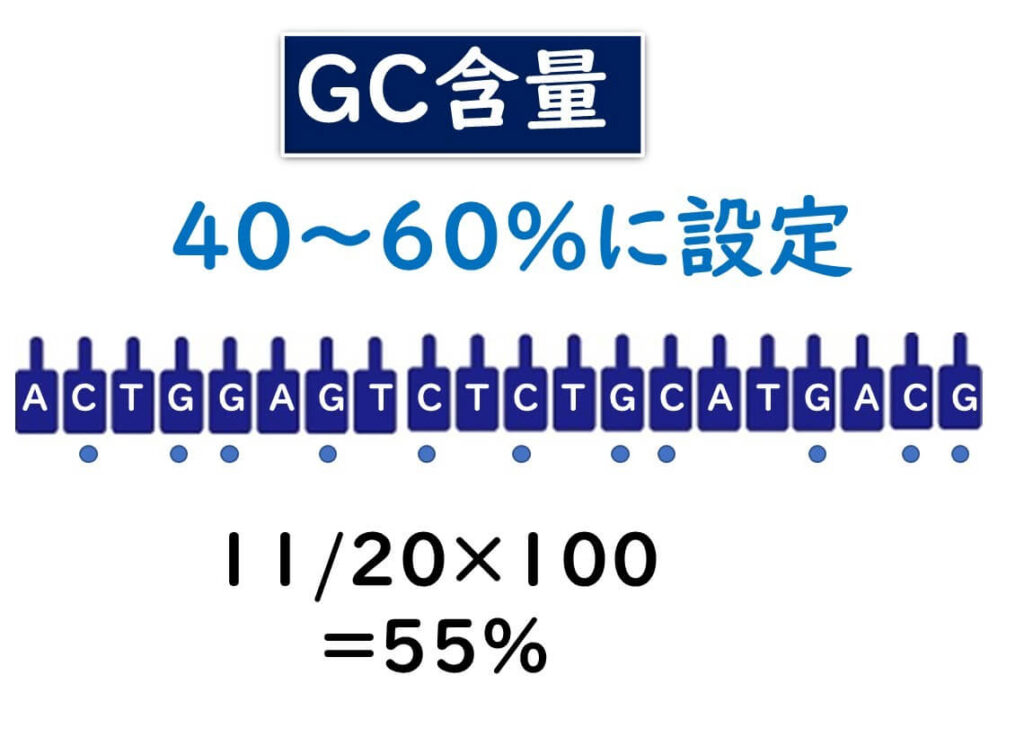
PCR法に用いるプライマーのGC含量は40~60%になるように設計する必要がある。
プライマーのGC含量の計算法
(GもしくはCの塩基数/プライマーの全塩基数)×100
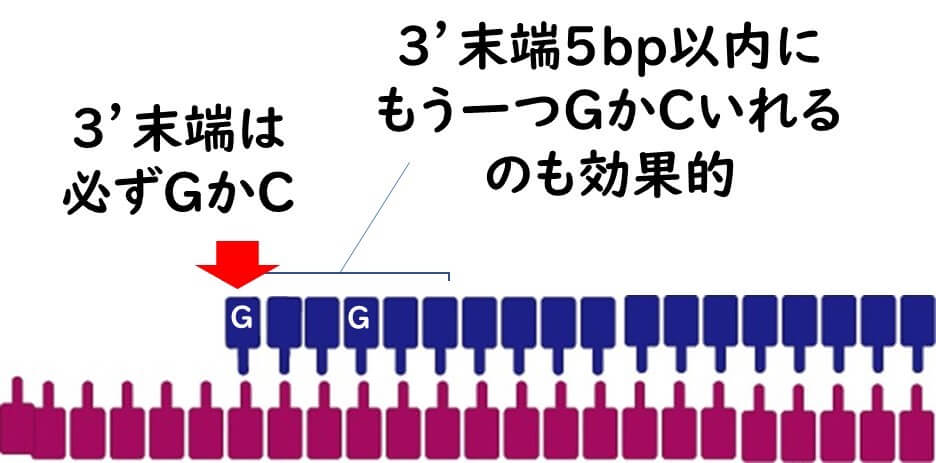
プライマーの3'末端の塩基はGもしくはCで終わるようにしなくてなならない。なぜなら、3’末端はDNAポリメラーゼが認識する場所だからだ。酵素の基質特異性により、この位置が揺らいでいてはDNAポリメラーゼは正しく伸長反応を始められない。上述したように GC間の結合は水素結合が3つあるのでAT間の結合より強力だ。したがって、 プライマーの3'末端の塩基はGもしくはC にすることにより、AやTが末端の場合より、末端の接着をしっかりと閉じさせることが出来る。
また、3’末端から数えて5塩基以内に、もう一つGもしくはC塩基が入ると、3’末端側がより安定となる。
ただし、3塩基以上は入れないほうがいい。3’側にGもしくはCが多すぎると、プライマー全体の配列の相補性が甘くても、3’側のみの安定性により非特異的にPCR増幅が起きる可能性が高くなるからだ。
一方、プライマーの5’側はAもしくはTが多くて、接着が甘い状態であっても、3‘側がしっかり接着していれば伸長反応は行われる。
とにかく、PCR反応のプライマー設計においては、プライマー全体のGC含量とともに、3‘側のGもしくはC塩基の配置に気を付けなくてなならない。

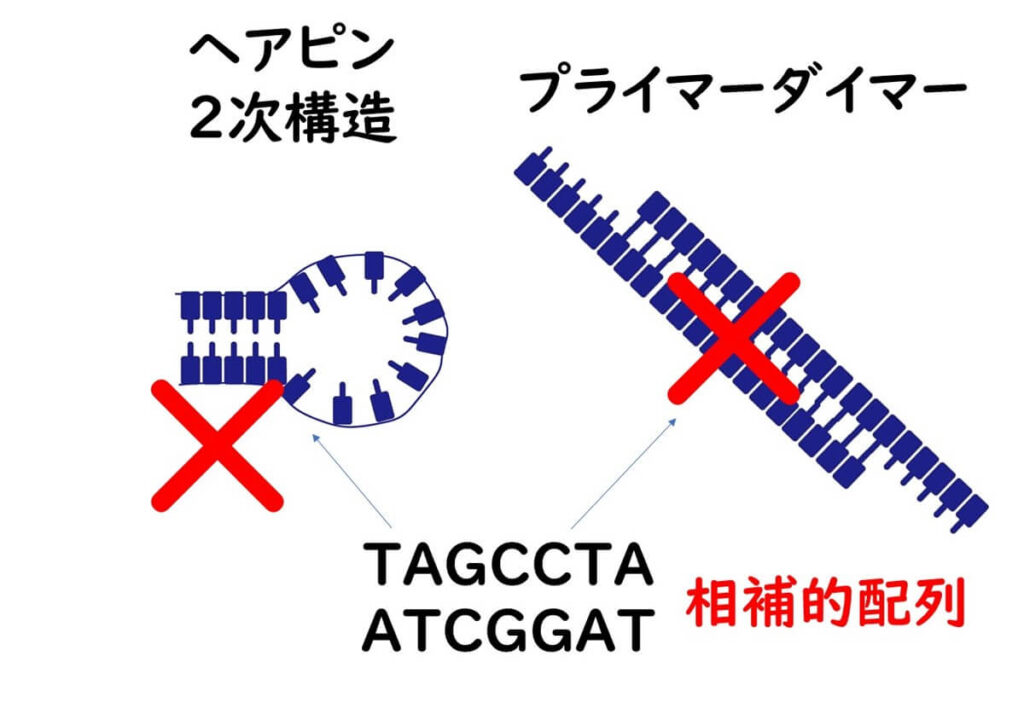
ヘアピンやプライマーダイマー
PCR反応のプライマー設計において、もう一つ気を配らなくてはならない点は、プライマー配列に起因する、次の2つのトラブルだ。
- ヘアピン2次構造
- プライマーダイマー
ヘアピン2次構造とは、プライマー内に相補的な連続配列が繰り返された要る場合だ。これらの相補配列がお互いにくっついて、下図のようにプライマーがヘアピン2次構造を形成してしまう。
また、プライマーダイマーとは、フォワードとリバースプライマー同士に連続した相補的配列がある場合である。この場合も、下図のようにプライマー同士が結合していまう。
ヘアピン2次構造、プライマーダイマーともに、プライマーは使い物にならなくなる。
遺伝子の両端認識しているに過ぎない
標的遺伝子の両端の遺伝子配列のみをその根拠として検出しているということを十分に理解しておく必要がある。

また特定の病原遺伝子を標的としてPCR検査を行う場合、標的遺伝子が増幅されたからと言って、その遺伝子が実際に細胞中で発現して病原性として機能しているとは限らない。
つまりPCR検査法は必ずしも盤石な論理的根拠に基づいた検出技術ではないという本質をまずは押さえておく必要がある。
生菌を検出できない
DNAは微生物細胞が死滅していても完全な形で残っている
遺伝子検査法の最大の弱点は、死菌のDNAも検出してしまうことである。 DNAは100°Cの煮沸では二本鎖を構成する水素結合が破壊され1本鎖になる。しかし、リン酸を介してデオキシリボースを結合している2重結合は切断されることはない。
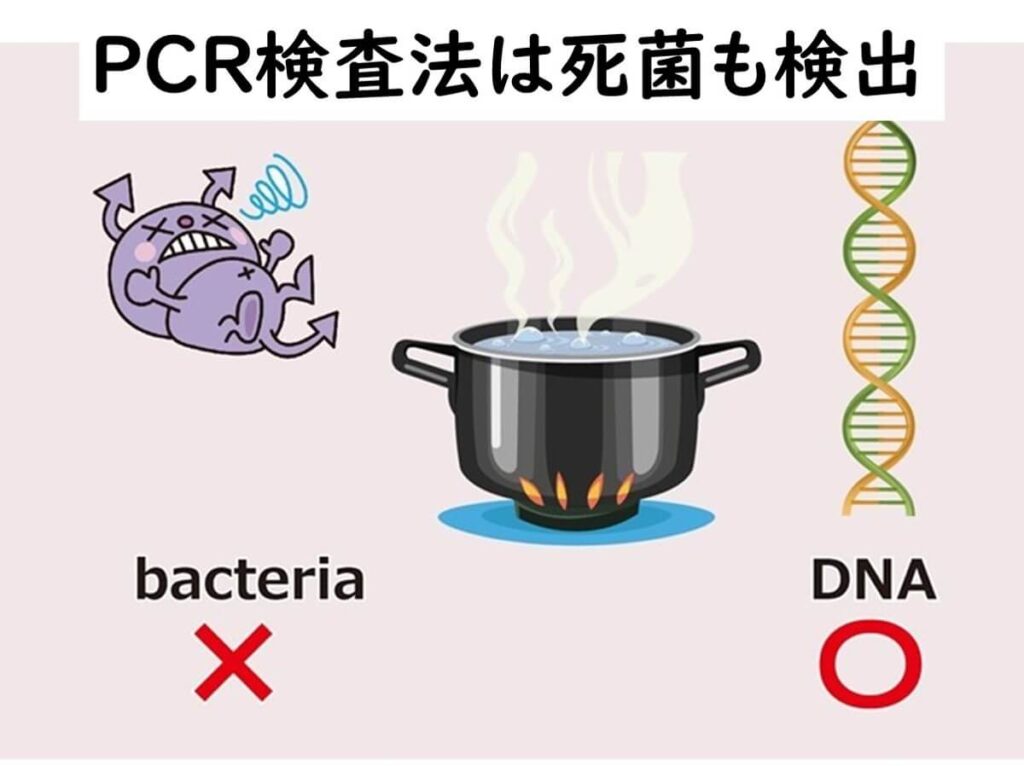
したがって、加熱などで微生物が死滅しても、遺伝子検査法の標的となる遺伝子配列はそのままは残存していることになる。したがって、PCR検査法で直接食品中の微生物を検査した場合は、得られる情報から微生物の生死は判定できない。

最近の研究で、Ethidium Monoazide (EMA) や Propidium Monoazide (PMA) のような、核酸共有結合色素を活用することにより、死菌を検出させないような工夫が試みられている。EMAやPMAは死んだ細菌の細胞膜を通過し、DNAに結合しPCR増幅を阻害する。したがって死菌の DNA からの遺伝子増幅が妨げられ、生菌の DNAのみから遺伝子増幅が行われる。しかしこのような方法によって得られる結果は、微生物細胞への損傷の種類によって異なる。PCR検査法においてこのような技術を導入することは可能であるが、法律的な側面などが関与する実用性においては、まだ道のりは遠い。
生菌検出問題は増菌培養で克服しているが
一方、食品検査の実用においては、遺伝子検査法の上述の欠点はあまり大きな問題となっていないのも事実である。その理由としてはPCR検査法の検出感度が低いために、食品 25 gに1細胞レベルでの 食中毒菌の検出感度を担保できないために、ほとんどの場合増菌培養が必要となるからである。増菌培養を行った後の培養液からの結果の判定は、生菌と判定できる。
注)なぜ、 PCR法では食品 25 gに1細胞レベルでの 食中毒菌の検出感度を担保できない なのかについては、下記別記事の中の【リアルタイムPCRと食品微生物検査】で説明しているので、ご覧ください。
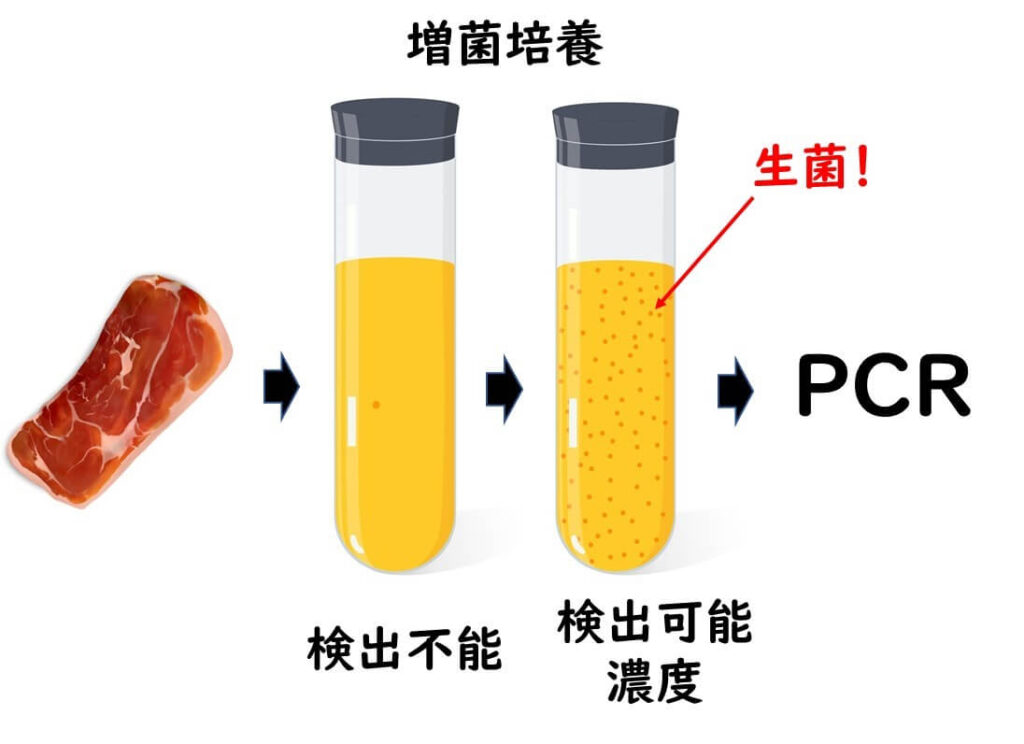
しかし一方で、このような増菌培養をかけると、25 gあたりに1だったのか、数百存在していたのかという定量的な情報は得られていないのが現状である。
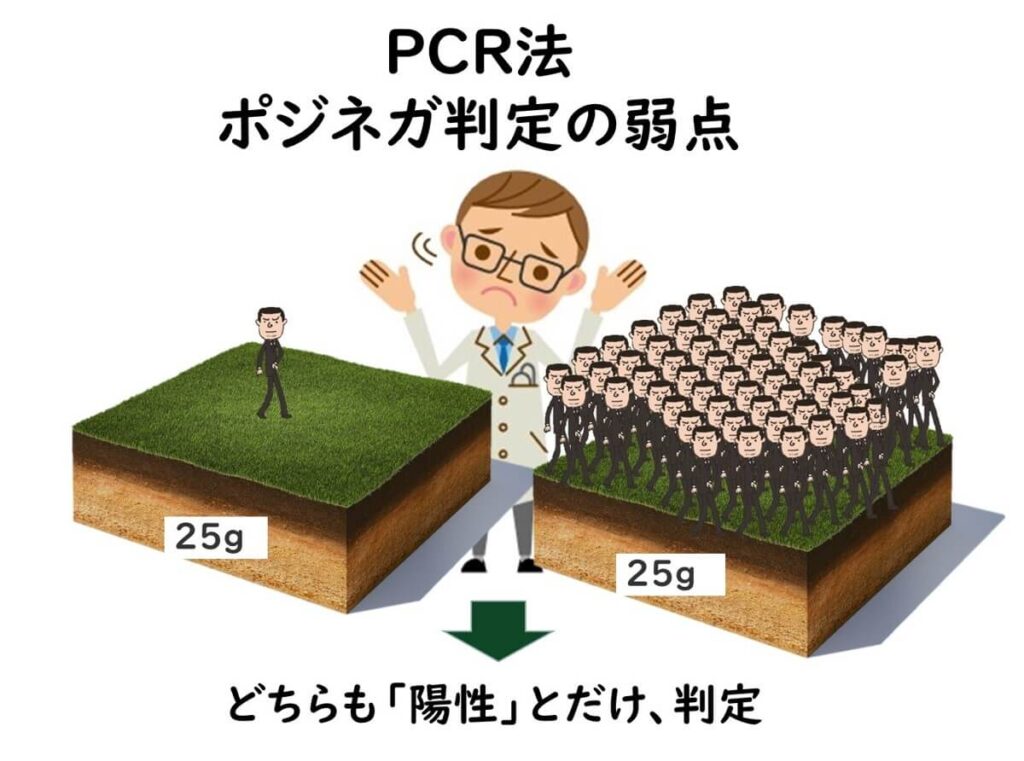
遺伝子検出 の盲点
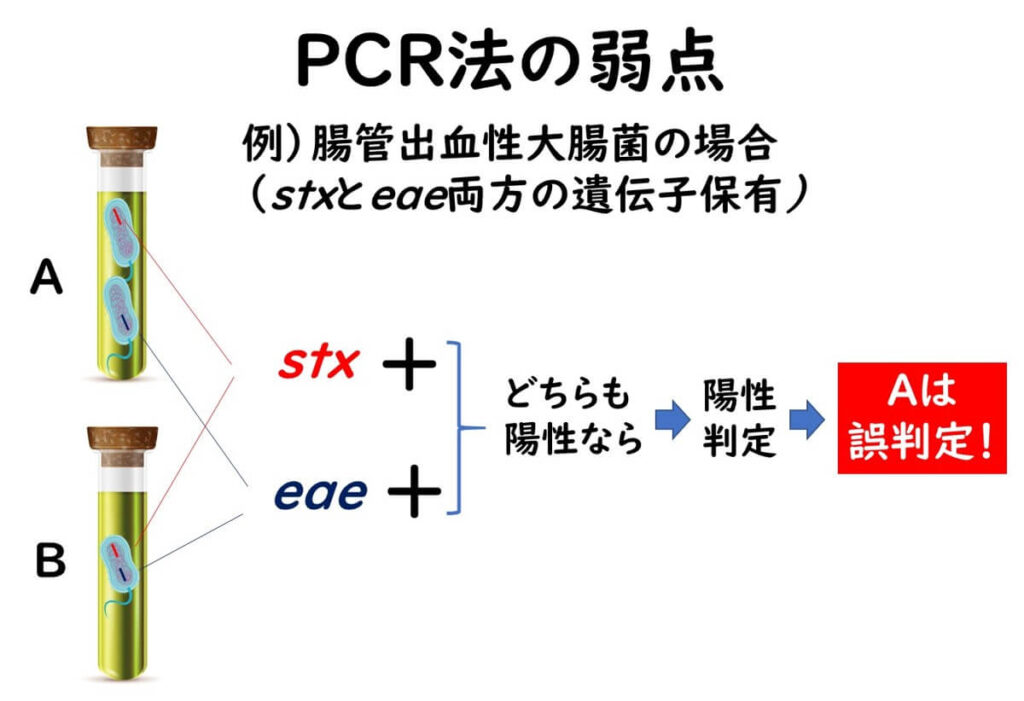
PCR検査法では、コロニーを単離せずも増菌培養から直接遺伝子を検出する。この手法の一つの盲点は、例えば、2つの遺伝子を標的として判定を行う病原性大腸菌などの場合である。増菌培養での判定では2つの遺伝子、stxとeaeの両方が判定されることによって潜在的陽性判定を行う。
しかし、この2つの遺伝子のどちらか一方のみを存在している別々の大腸菌がその増菌培養に存在している場合などは、誤判定となる。この場合その後の培養法では標的とする病原性大腸菌を検出できないことになる。
実際に現在、国際的なISO法でも国内の公定法でも腸管出血性大腸菌の検査においては、PCR法での陽性になっても、その後の培養では、多数のサンプルで陰性となる問題を抱えている。その主因と考えられるのがここで述べた遺伝子検出の盲点に起因していると考えられている。
増菌培養時の注意事項
PCR検出にはとにかく増菌培養で12回分裂を行わせればよい
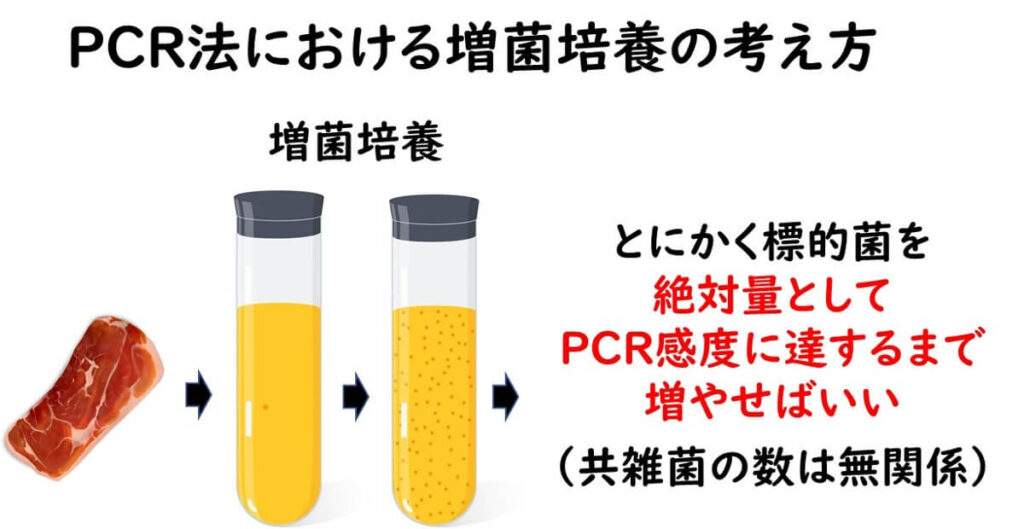
培養法では、雑多な共雑菌が標的菌とともに存在している食品から、いかに、標的菌のみの割合を上げていくかがポイントとなる。そうしないと、最終的な検出平板培地で標的菌をピックアップできない。したがって、選択剤をいれた選択増菌培養が不可欠となる。

一方、PCR検査法では、培養法と異なり増菌培養中に標的菌数が絶対数として一定の数にさえ増殖させれば、検出が可能となる。この点は背景の雑菌との相対的な割合が検出の是非を決める培養法とは異なる。
この意味で遺伝子検査法の増菌培養の仕組みより単純である。培養法のように、選択剤を培地に添加しなくても、場合によってはペプトン培地などのような増菌培養だけでも検出ができる場合も多い。
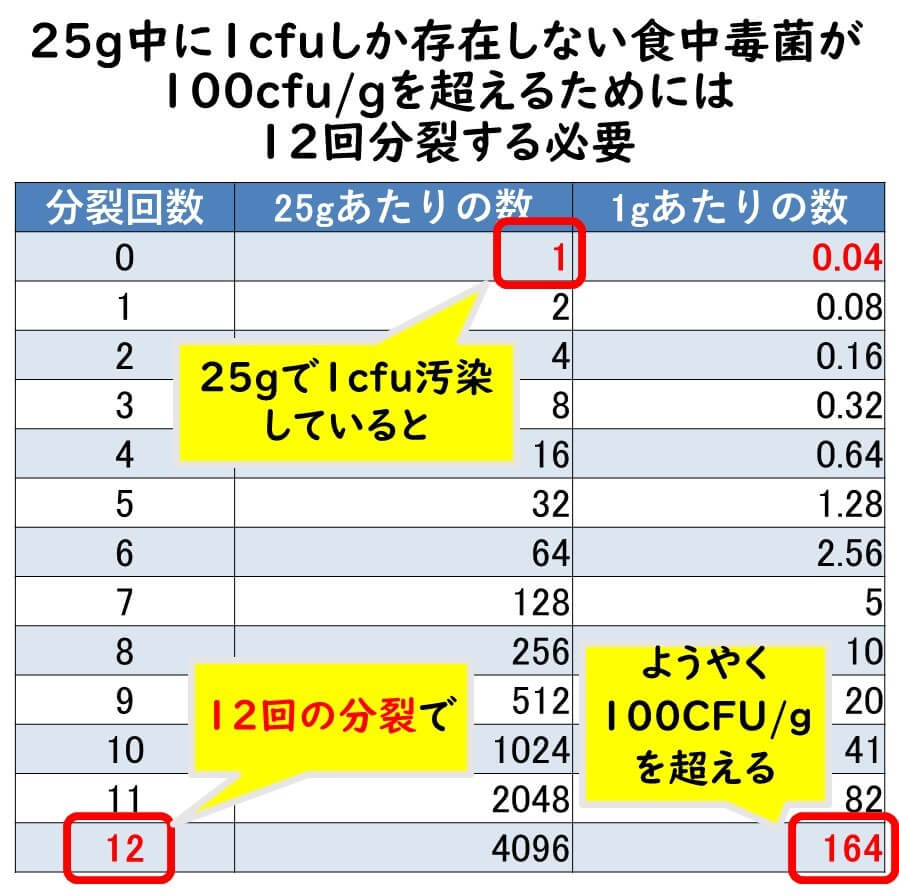
一般的にPCR法の検出感度は食品中で100cfu/g注)である。したがって、25g中に 1細胞存在する標的微生物が遺伝子検査法の検出感度までに増えるためには大雑把に言えば、とにかく12回の分裂させれば良いということになる。
注)なぜ、PCR法の感度が 食品中で100cfu/g なのかについては、下記別記事の中の【リアルタイムPCRと食品微生物検査】で説明しているので、ご覧ください。
PCR法の増菌培養時の盲点
上述したように、PCR検出では、標的菌をPCR感度に達するまで、すなわち、12回分裂させさえすればよい。これだけの目的なら、増菌培地に選択剤を入れなくてもいいので、増殖するTSB培地やユニバーサル培養のようなペプトン液体培地を用いる場合も多い。
しかし、ここでも、盲点がある。
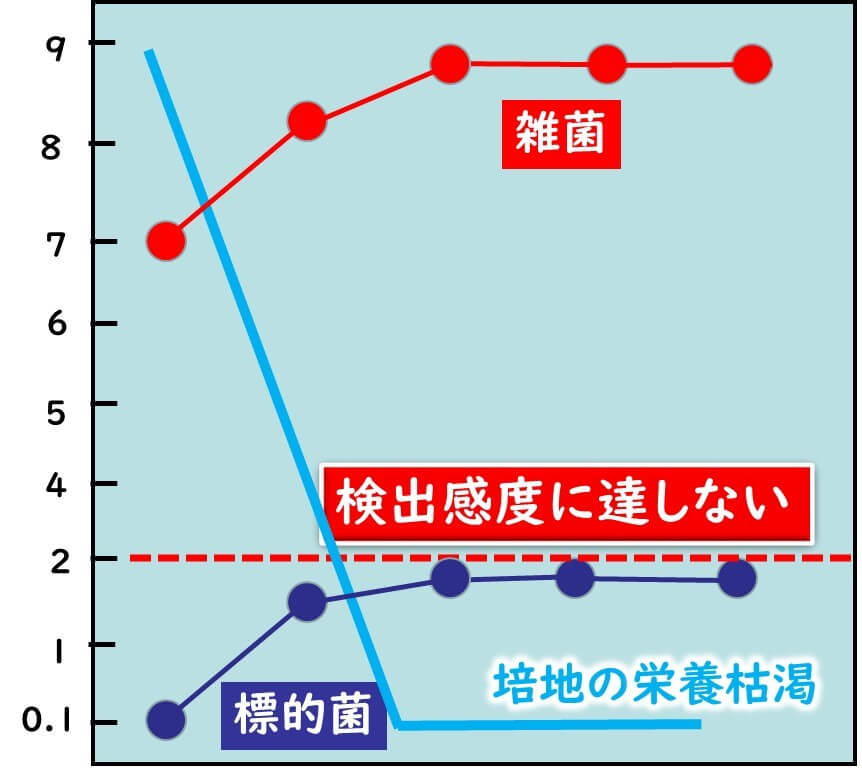
増菌培養での12回の分裂を阻害する要因もある。
その一つは、香辛料など、そもそも標的菌の増菌培養中での増殖を阻害する場合である。

もう一つは、かいわれ大根などのように共雑細菌の数が多すぎるために、競合により増菌培養で標的菌の増殖が十分行われない場合である。PCR検査法を用いる場合には、これらの2つの場合については特に注意が必要である。

遺伝子増幅時の注意事項
遺伝子増幅を阻害する要因(偽陰性)
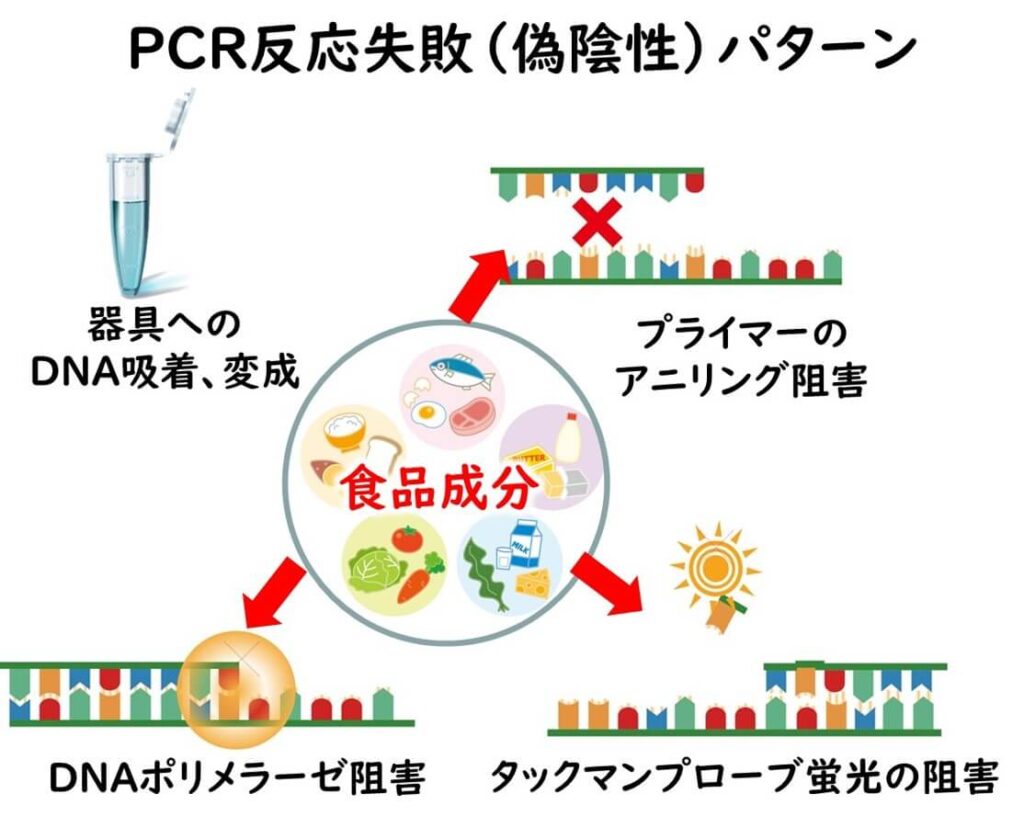
遺伝子検査における遺伝子の抽出および増幅過程において食品成分由来の成分が遺伝子の増幅を阻害する場合もある。
遺伝子増幅段階での阻害が起きる要因としては、
1)遺伝子の抽出の際に標的DNAが容器などに吸着して変成してしまうケース
2) PCR などの増幅過程におけるプライマーが標的遺伝子に接着するのを妨げてしまうケース
3) DNA ポリメラーゼの酵素作用を阻害してしまうケース、
4)タックマンなど蛍光プローブを用いる場合は、蛍光発色を阻害してしまうケース
などに大別できる。
食品微生物学分野においてPCR反応によって食品から特定の微生物の検出を行おうとする場合、食品中に含まれるPCR反応の阻害物質の影響を考慮する必要がある。PCR反応が陰性となった場合、標的とする微生物が存在していなかったのか、あるいは食品中に含まれるPCR反応阻害物質の影響により陽性反応にならなかったのかについての区別が困難となる。
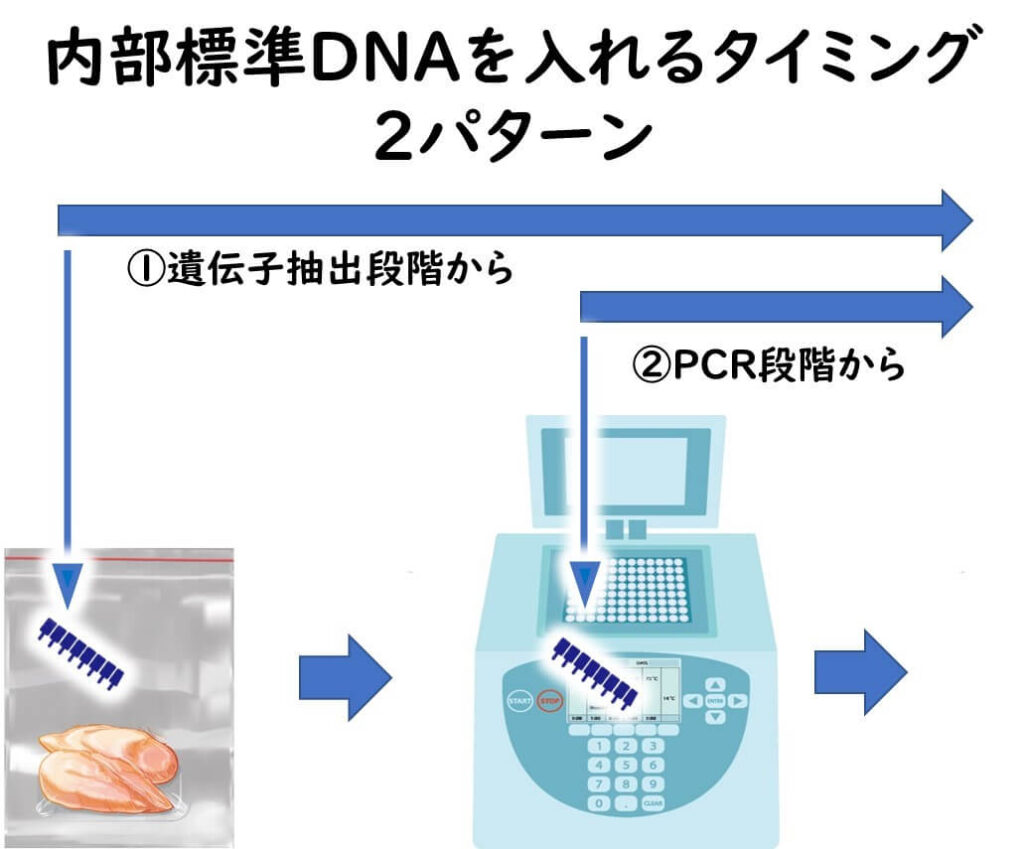
そこで、このような区別を明確にするために、あらかじめ配列のわかっている遺伝子断片をPCR反応の中に組み込んでおく。この断片のことを 内部標準(internal positive control:IPC)を遺伝子 と呼ぶ。 内部標準 遺伝子の増幅が行われていれば、少なくとも食品中のPCR反応阻害物質がPCR反応を阻害していないと判定することができる。

内部標準には、食品の抽出の段階から加えるものと、遺伝子の増幅時にのみ加えるものとがある。一般的には遺伝子断片を加える場合が多いが、理想的には、食品そのものに対象となる標準菌株そのものを接種して、対象とする食品の遺伝子の抽出の段階から検証しておくのが理想的である。
遺伝子の混入に注意(擬陽性)
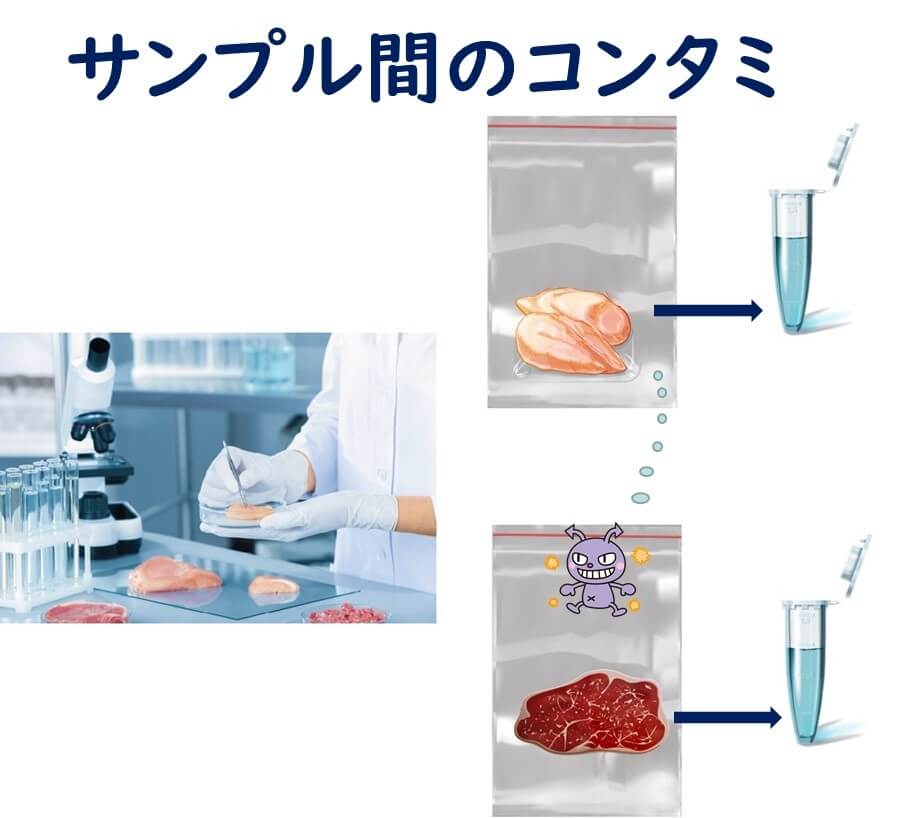
次にPCR査におけに擬陽性を引き起こす要因について簡単にまとめてみる。偽陽性はサンプル中に本来存在していない遺伝子がサンプルに誤って混入することによって起きる。その原因を大別すると、
1)異なるサンプル間での遺伝子の混入
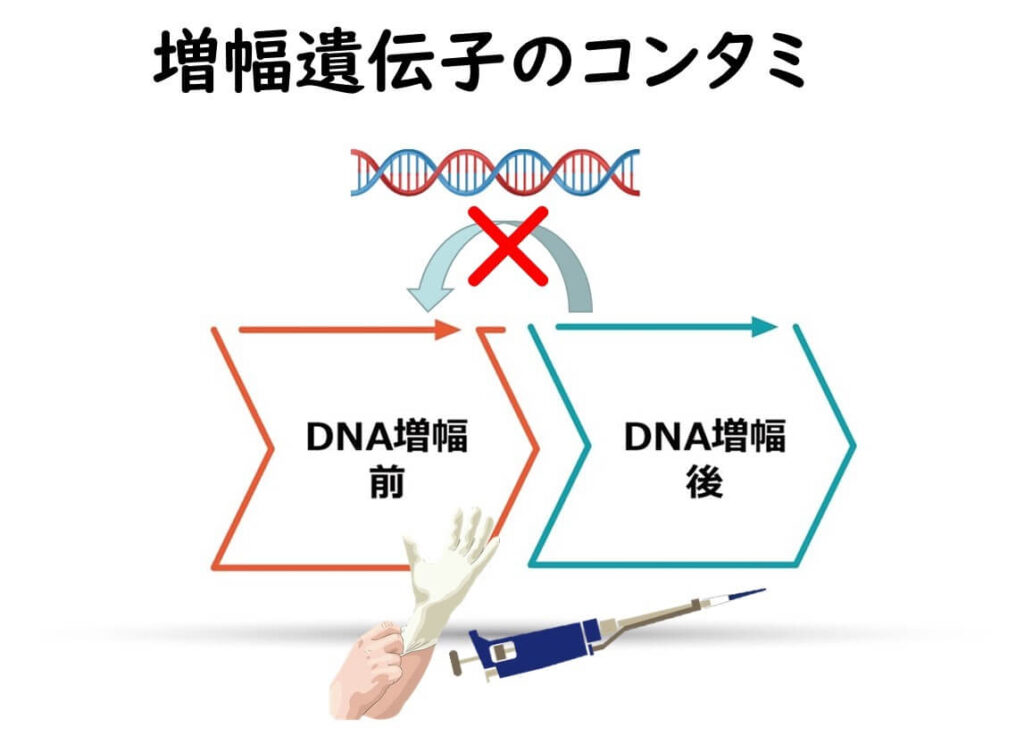
2)遺伝子を増幅した分析用のサンプルから新たなサンプルへの混入
に整理することができる。

これらの混入を防ぐためには、まずは実験室の ゾーン区分が必要となる。
理想的には遺伝子を増幅する前のサンプルを調製する実験室と、遺伝子を増幅した後の分析をする実験室は分けるのが望ましい。これができない場合は、同じ実験室内でも実験台に明確なゾーン区分をすることが必要となる。これらの区分間においてピペット、ガラス器具、手袋、白衣などを共有しないことが重要となる。
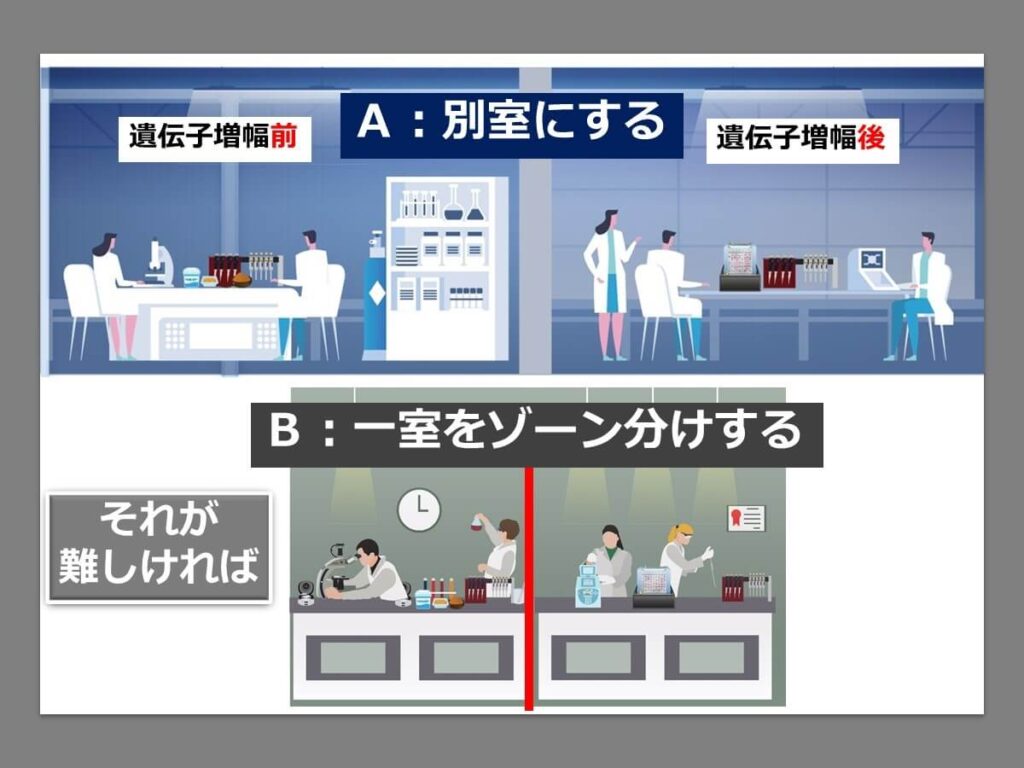
白衣なども基本的に着替えの方が望ましいが、小さな実験室でこれが不可能な場合は、少なくとも実験を行う直前に実験台を次亜塩素酸ナトリウムなどでの拭き掃除により清掃する習慣を身につける必要がある。
またピペット操作などについても落ち着いてゆっくりと行いサンプルを飛び散らしてエアロゾルが空気中に飛散しないような訓練も必要となる。このような注意事項は日頃から訓練をして身につけておく必要があるだろう。

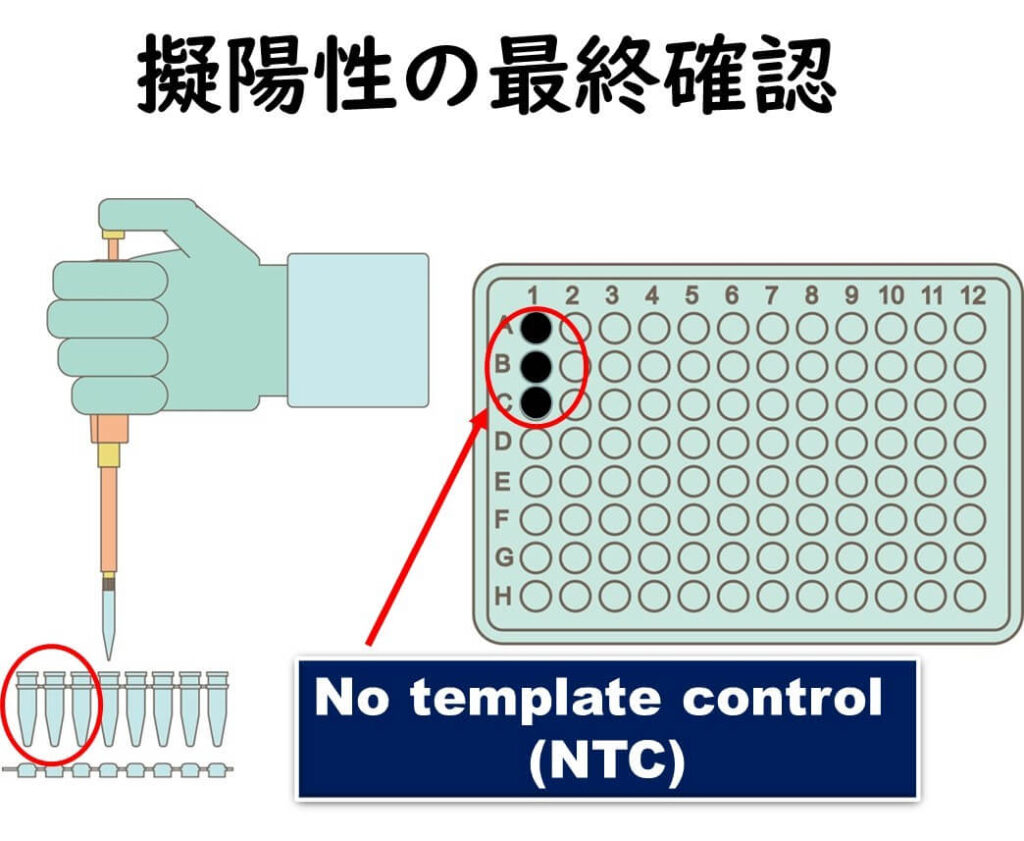
このように日頃から遺伝子のコンタミネーションの注意を払った上で、 実際の分析においては、増幅の対象とする遺伝子を一切含まない No template Control(ネガティブコントロール)を実験系に組み込んでおくことが必要となる。このようなサンプルにおいて仮に陽性反応が出てしまうことが頻繁に起きるようであれば実験室の管理を根本的に見直す必要がある。
食品微生物検査でPCR法の期待される用途
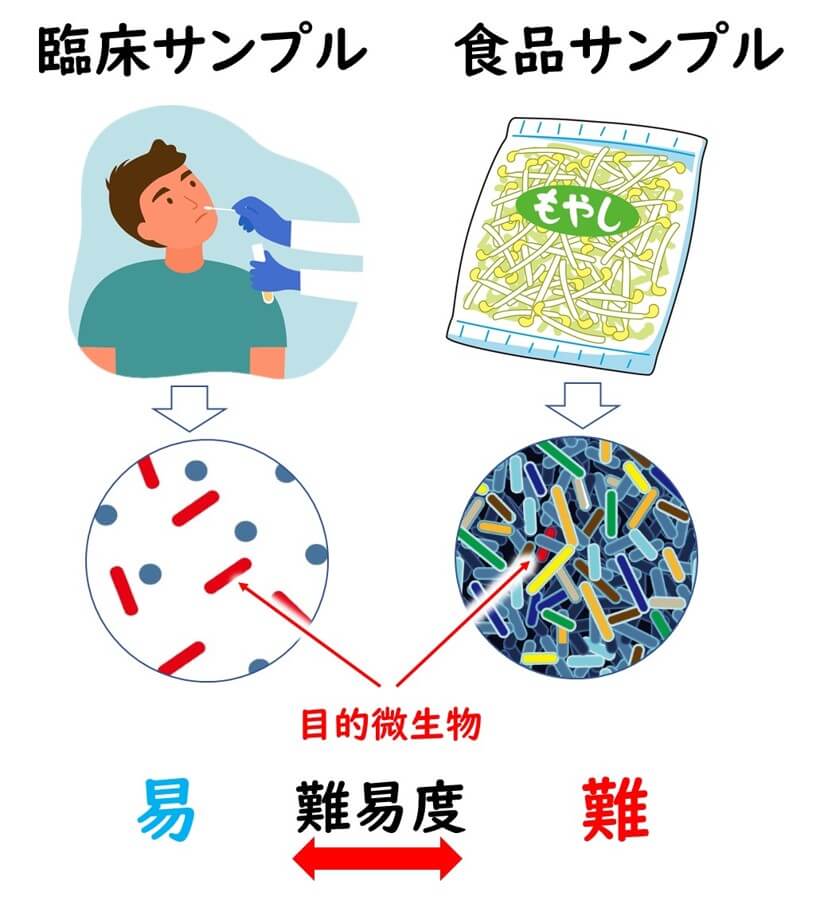
食品からの微生物検出は臨床微生物検出より難易度が高い
2020年に新型コロナウィルスが流行してPCR法という言葉はすっかりなじみになった。
ところで、PCR法による病原菌の検出でも、臨床サンプルと食品サンプルからの検出では、難易度が異なる。
一般的には、食品サンプルのほうが難易度が高い。
その理由は臨床サンプルと食品サンプルのバックグラウンドに存在する共存微生物の差である。臨床サンプルで、特に、血液サンプル、鼻やのどの拭き取りサンプルなどの場合、そこに共存している微生物と種類はそれほど多いわけではない。 一方、食品サンプルの場合、雑多な微生物の中でわずか25 g中1 cfu 存在する標的菌を検出する必要がある。
従って PCR のプライマーの設計の精度はもとより、 PCR 反応の前ステップしてての選択増菌培養等の工夫などが食品の場合には求められる。
バリデーションのないPCR検出は信用できない
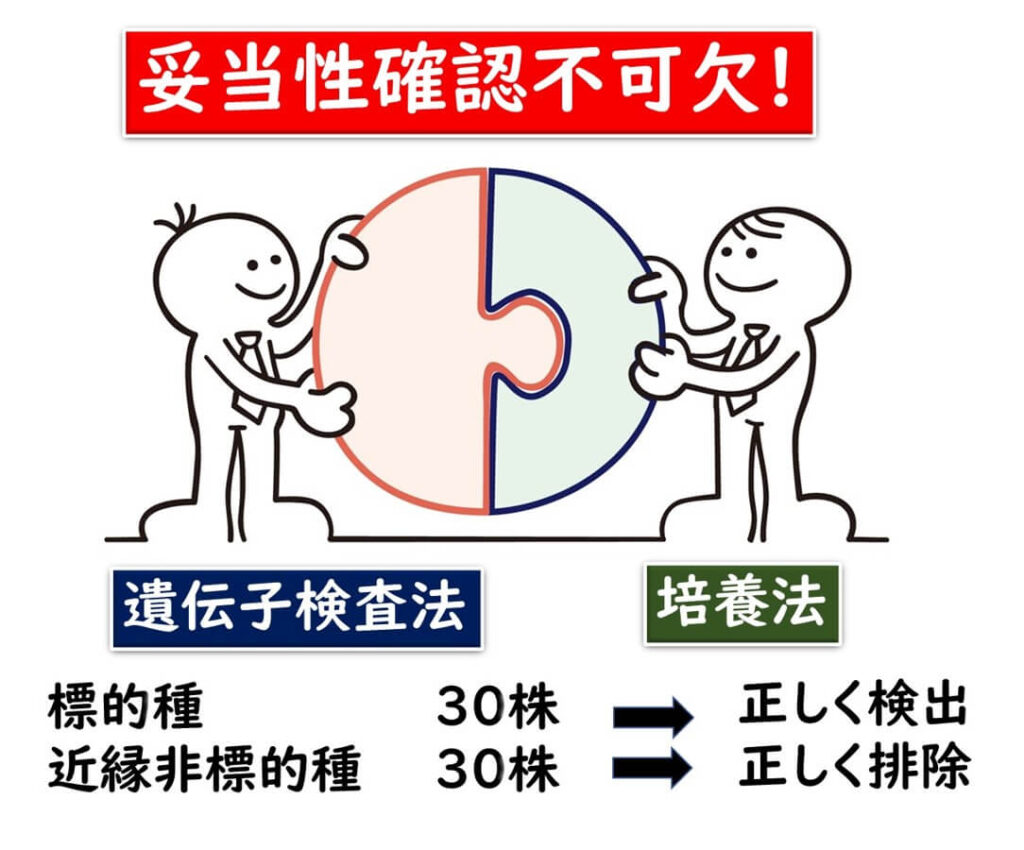
本記事で説明したように、食品の微生物検査法としてのPCR検査法には本質的に危うさが包含されている。だからこそ、しっかりとした妥当性確認のプロセスが必要となる。そのためには、多数の株を用いて妥当性確認の試験を行う必要がある。
例えば、 ISO(ISO 16140:2003)やAOACなどの国際標準法の妥当性確認のプロセスでは、目的とする標的株と標的から外すべき細菌株について次の基準が設定されている。
- 標的種の株:30株
- 標的種の近縁種(PCRでは標的外の種)の株:30株
このように、それぞれ最低30株を用いて正確に識別できることの証明が求められる(対象菌によっては50株)。
このような妥当性確認は、遺伝子検査法においては極めて重要なプロセスとなる。言い換えると遺伝子検査法においては妥当性確認の行われてない方法に関しては、その結果の信用性が乏しいと考えておくべきである。
筆者はInternational of Food Microbiologyの編集に2012年以降携わっているが、新たなPCRプライマーを用いた微生物の検出法の提案に関する論文が毎週多数投稿される。これらを精査すると、上記のような30株:30株の検証を行っていない場合がほとんどである。著者らの任意の判断で数株から10株程度で特異性があるとしている場合が多い。もちろん、これらの論文は、 30株:30株の検証 を行っていないだけの理由では必ずしもリジェクトになならない。アカデミアの論文では、新規性やアイデアを重視して掲載する場合も多いからだ。また、他のジャーナルなどでは、厳密なvalidation無しで出版されている論文も多数ある。
すなわち、インパクトファクターの高い査読付き国際論文雑誌に掲載されていることと、国際的な認証機関でvalidationが行われていることは別問題であることに留意する必要がある。立派なジャーナル掲載されている論文の方法だからといって、その精度を鵜呑みにしてはならない。
食品微生物検査委にPCR法を採用する場合は、 ISOやAOAC などのvalidationをクリアしているかに、十分留意する必要がある。
以上、本記事では食品から微生物を PCR 検出する場合の各種の注意事項や留意点について述べてきた PCR 法は使い方によっては便利であるがいくつかの落とし穴やリスクがあるということを十分理解した上で使うのがよいだろう。

PCR検査の作業スケジュール
最後に、食品の微生物検査をPCR法で行う場合の作業スケジュールイメージについても簡単に記しておく。
食品製造流通現場でのルーティン検査では、通常、25g当たりに食中毒菌が1細胞存在しているか否かを問題とする場合が多い。つまり、対照試料中の標的遺伝子量が圧倒的に少ない。従って、いかにPCR検出法といえども、一度食品中の食中毒菌をある程度の量まで増やす(増菌培養)ステップが必要となる。しかし、培養法のように長時間を必要とせず、通常数時間で十分な場合が多い。従って、サンプル調製と培養時間も含めて一日の作業時間で分析をすべて終えることももちろん可能であるが、現実問題として、朝9時から夕方5時の勤務時間を考慮すると、次のような作業手順が最も自然で無理がないのではないだろうか。
1日目:夕方食品を増菌培地と混合し(25gを225mlに)ストマッカーにかけ、これをそのまま37℃のインキュベータに入れて帰宅する。
2日目:朝9時に出勤したら直ちに分析を開始し、DNAの抽出、PCR増幅、結果の判定を午前中一杯で終了する。
以上のような手順で、毎日食品サンプルについて食中毒菌の混入の有無が判定できるので、HACCP方式を導入した食品製造現場で原料や途中加工製品の微生物学的品質の迅速判定に大きな力を発揮することが可能である。

